Chemistry Review – Part 2
2. Acids and Bases
Acid-base chemistry is critically important in hydrometallurgy. Most processes involve reactions where acids or bases are used, and with varying pH in different parts. For example, many leaching processes for the dissolution of minerals use sulfuric acid. Dissolution of bauxite ores for aluminum processing require strong base.
Note: As noted in Chemistry Review Part 1, the proton is present in water as H3O+, never as H+ per se. For convenience we often designate it as H+, but it is understood to be actually present as hydronium ion, H3O+.
2.1 Acid-Base Theories
1. Arrhenius Acids and Bases.
Acid-base behaviour was first noted for aqueous solutions. The first and simplest concept was the Arrhenius theory. It stated that an acid was anything that had an ionizable proton attached to it, and a base was anything with an ionizable OH- group. Examples include:
H2SO4, CH3CO2H, (H-NH3)2SO4 i.e. (NH4)2SO4, etc. (acids)
NaOH, Mg(OH)2, etc. (bases)
The acid protons/basic OH groups are indicated in bold. This concept did no go far enough. It did not recognize that many acids and bases react with water to form H+ or OH-, even though they may not contain ionizable -H or -OH groups. By the Arrhenius theory NH3 would not be a base, yet it does release OH- into solution when it dissolves in water; and by the same token CO2 would not be and acid, yet it results in H+ being formed in solution:
\[ \ce{NH3_{(g)} + H2O_{(l)} = NH4+_{(aq)} + OH-_{(aq)}} \tag{54}\]
\[ \ce{CO2_{(g)} + H2O_{(l)} = HCO3-_{(aq)} + H+_{(aq)}} \tag{55}\]
2. Lowry and Brønsted Acids and Bases.
Lowry and Brønsted extended the acid-base concept to say that an acid is any substance that can cause a proton to form in water (it does not necessarily have to have one bonded to itself to begin with) and a base is one that can accept a proton. In the above reactions NH3 is accepting a proton from H2O and CO2 is reacting with water to donate a proton. Hence NH3 is acting as a base and CO2 as an acid.
3. Conjugate Acids and Bases.
In reaction (54) note that NH3 is the base and H2O is acting as an acid; water gives up an H+ to NH3. At the same time, NH4+ is an acid and OH- is a base; they can react according to the reverse of reaction (54). This leads to the important idea that acid-base reactions in water follow the form:
\[ \ce{Acid_1 + Base_1 = Acid_2 + Base_2} \tag{55}\]
For a reaction of an acid we can write a general reaction,
\[ \ce{HA + H2O_{(l)} = H3O+_{(aq)} + A-_{(aq)}} \tag{56}\]
HA and A- differ from each other by only a proton. Likewise H2O and H3O+. HA and A- are said to be conjugate acid-base pairs. For a base we write the general reaction,
[ \ce{B + H2O_{(l)} = BH+_{(aq)} + OH-_{(aq)}} \tag{57}\]
B and BH+ are conjugate acid-base pairs. This will become important when we consider buffers. At this stage the key concept is that each acid has its corresponding base, and that they differ by only one H+.
4. Lewis Theory of Acids and Bases.
The third theory of acids and bases is the Lewis theory. It extends acid-base behaviour to any type of solvent, and it recognizes that proton acidity in water is only one class of a more general acid-base phenomenon. In this theory a base is anything that can donate a pair of electrons, and an acid is anything that can accept a pair of electrons. Considering reaction (54) ammonia has a lone pair of electrons that is not used in bonding to the hydrogens. Recall that ammonia is a group Vb element. It has five valence electrons. Three of them are involved in single electron-pair bonds with the hydrogens. Two remain unused in bonding. When NH3 reacts with water it abstracts a proton, H+. A new N-H bond is formed. It is an electron pair bond, but both electrons came from the nitrogen. Hence H+ is accepting a pair of electrons and NH3 is donating a pair of electrons.
Extending these ideas, H2O is a divalent oxygen compound. Oxygen is a group VIb element with six valence electrons. Hence two pairs of electrons remain on the oxygen. When water coordinates to a metal ion, e.g. [Fe(H2O)6]+2, each Fe+2 forms six electron pair bonds, one with each water. But both electrons come from the water. Water is acting as a base, in the Lewis sense; Fe+3 is acting as an acid. A great many complexes involve just this sort of acid-base interaction. This is certainly true of virtually all the complexes of interest in hydrometallurgy. But, ligands such as CN- can extend this behaviour to yet another degree. Cyanide can be depicted as -C≡N, where C and N are joined by a triple bond (three electron-pair bonds). With the 1- charge formally on the carbon atom, tetravalent carbon (NB, group IVb) has a pair of electrons on it. These are donated to a metal ion in the acid-base manner just described. The nature of the multiple bond between C and N, however, means that there are empty “orbitals” (energy levels where electrons could reside, but do not) that can accept electrons from the metal ion. Now this additional acid-base interaction is the other way around; from the metal ion to the ligands. This can greatly strengthen the metal-ligand bond, and as a result many cyanide complexes are exceedingly stable. This has great significance for hydrometallurgy. [Au(CN)2]- is an unusually stable complex of Au(I). This allows for the processing of almost all gold ores. Cyanide is used to dissolve solid gold from the ore to form the complex above. This in turn can be recovered from the solution by various means, eventually facilitating the production of gold metal. Without this chemistry, a great deal of the world’s gold production would not be possible.
2.2 Acid-Base Equilibrium Constants.
Acid-base reaction equilibrium constants are set up in exactly the same way as outlined previously. The acid dissociation equilibrium constant is designated Ka. It has the general form:
\[ \ce{HA_{(aq)} + H2O_{(l)} = H3O+_{(aq)} + A-_{(aq)}} \quad K_a = \frac{\ce{[H3O+][A-]}}{\ce{[HA]}} \tag{58}\]
and equivalently,
\[ \ce{HA_{(aq)} = H+_{(aq)} + A-_{(aq)}} \quad K_a = \frac{\ce{[H+][A-]}}{\ce{[HA]}} \tag{59}\]
That H+ is actually present as H3O+ is implicit in the latter. For an anionic base (e.g. HS-, F-, CN- etc.) we can write:
\[ \ce{A-_{(aq)} + H2O_{(l)} = HA_{(aq)} + OH-_{(aq)}} \quad K_b = \frac{\ce{[HA][OH-]}}{\ce{[A-]}} \tag{60}\]
For neutral bases (e.g. NH3) we write:
\[ \ce{B_{(aq)} + H2O_{(l)} = BH+_{(aq)} + OH-_{(aq)}} \quad K_b = \frac{\ce{[BH+][OH-]}}{\ce{[B]}} \tag{61}\]
The form is exactly the same! These are only schematic ways of expressing the same base behaviour.
2.3 Strong and Weak acids and Bases.
As previously, the larger the value of Ka or Kb, the more it dissociates, i.e. the further the reaction proceeds to the right, and the stronger the acid or base. In the past a strong acid was considered to completely ionize in solution, meaning that none of the parent acid can be detected in solution. Then for a strong acid HY would have an undefined Ka, since [HY] would be zero:
\[ \ce{HY -> H+_{(aq)} + Y-_{(aq)}} \tag{62}\]
\[ K_a = \frac{\ce{[H+][Y-]}}{\ce{[HY]}} \tag{63}\]
However, it has been found that even the strongest acids do not completely dissociate, as one might expect. An example is HCl. Indeed in water very little molecular HCl persists. The compound almost completely dissociates into H+ and Cl-. Since [HCl] is exceedingly small, Ka is large, i.e. ~107. In a 1 M HCl solution the concentration of HCl molecules would be only ~10-7 M.
There are not very many strong acids and bases. A list of the main strong acids is provided below.
Strong acids:
- H2SO4 (first dissociation only, i.e. H2SO4 = H+ + HSO4-; Ka ~1000)
- H2SeO4 (first dissociation only; Ka ~100)
- HCl (also HBr, Ka ~109; HI, Ka ~ 1010)
- HNO3 (nitric acid, Ka ~ 25)
- HClO3 (chloric acid, Ka ~10; not commonly used)
- HClO4 (among the very strongest acids, Ka ~1010; powerful oxidant; may react violently with reducing matter)
- H2SiF6 (important in lead processing)
- HBF4 (not commonly used)
We will consider an acid to be “strong” if Ka exceeds 10. The first dissociation of H2SO4 at room temperature is taken to be complete. The hydrohalic acids and the oxoacids are known as mineral acids. Strong bases almost completely dissociate in water to form hydroxide ions. While pure strong acids are largely molecular compounds (not ionic), strong bases are all ionic solids.
Strong bases:
- NaOH (also KOH and other alkali metal hydroxides)
- M2O (alkali metal oxides; O2- ion does not survive in water)
\[ \ce{M2O_{(s)} + H2O_{(l)} = 2M+_{(aq)} + 2OH-_{(aq)}} \tag{64}\]
We can write the dissociation of HCl as follows:
\[ \ce{HCl_{(aq)} + H2O_{(l)} = H3O+_{(aq)} + Cl-_{(aq)}} \tag{65}\]
Clearly Cl- is the conjugate base of HCl. However, since HCl is such a strong acid, Cl- must be an exceedingly weak base, so weak that for the reaction:
\[ \ce{Cl-_{(aq)} + H2O_{(l)} = OH-_{(aq)} + HCl_{(aq)}} \tag{66}\]
the equilibrium constant is ~10-21. That Cl- is such a weak base is implied by the fact that HCl is such a strong acid.
Most acids and bases are weak; they dissociate incompletely. Roughly speaking, an acid can be considered to be weak if Ka < 10. A few of the important weak acids and bases relevant to hydrometallurgy are listed below.
| Table 2.1 - Weak Acids & Bases | |
|---|---|
| XXX | XXX |
| H2O + H2O = H3O⁺ + OH⁻ | water self-ionization |
| NH₄ + H2O = H3O+ + NH3 | ammonium/ammonia |
| HCN + H2O = H3O+ + CN⁻ | hydrogen cyanide/cyanide |
| H2S + H2O = H3O+ + HS- | hydrogen sulfide/bisulfide (or hydrosulfide) |
| HS- + H2O = H33O+ + S2- | bisulfide/sulfide |
| CO2aq + H2O = H2CO3 | carbon dioxide forming carbonic acid |
| H2CO3 + H2O = H3O+ + HCO3- | carbonic acid/bicarbonate |
| CO2 aq + H2O = H3O+ + HCO3⁻ | carbon dioxide/bicarbonate |
| HCO3- + H2O = H33O+ + CO32- | bicarbonate/carbonate |
| SO2 + H2O = SO2 aq | (sometimes written as H2SO3, “sulfurous acid,” however H2SO3 is not known to exist.) |
| SO2 aq + 2H2O = H3 O+ + HSO3- | sulfur dioxide/bisulfite |
| HSO3- + H2O = H3O+ + SO32- | bisulfite/sulfite |
| HSO4- + H2O = H3O+ + SO42- | bisulfate/sulfate (the second dissociation of H2SO4 is weak) |
| HF + H2O = H3O+ + F- | hydrofluoric acid/fluoride (of the hydrohalic acids, only HF is not a strong acid) |
Carbon dioxide and sulfur dioxide are somewhat more complicated. Both dissolve in water as the molecules, as written above. Carbon dioxide manifests acidity in two ways:
\[ \ce{CO2_{(aq)} + H2O_{(l)} = HCO3-_{(aq)} + H+_{(aq)}} \tag{67}\]
\[ \ce{CO2_{(aq)} + H2O_{(l)} = H2CO3_{(aq)}} \tag{68}\]
and H2CO3 itself also dissociates:
\[ \ce{H2CO3_{(aq)} = H+_{(aq)} + HCO3-_{(aq)}} \tag{69}\]
Sulfur dioxide does not appear to form H2SO3 to any detectable extent thus far:
\[ \ce{SO2_{(aq)} + H2O_{(l)} = HSO3-_{(aq)} + H+_{(aq)}} \tag{70}\]
\[ \ce{HSO3-_{(aq)} = SO3^2-_{(aq)} + H+_{(aq)}} \tag{71}\]
Note: Each acid listed above has its corresponding conjugate base. For instance, the conjugate base of H2S is HS- (not S2-), and the conjugate base of HS- is S2- (not HS-); again, conjugate acid base pairs differ by only one H+, although the addition or loss of water may also be included, as in CO2 (acid) and HCO3- (conjugate base). Note too that A species may acts as a base or as an acid, depending on the reaction (e.g. HCO3- and HSO3-). Such species are said to be amphoteric.
2.4 Water and Sulfuric Acid.
As mentioned earlier, water is a weak acid and a weak base. Thus it is also amphoteric. An amphoteric compound is necessarily only weakly acidic and weakly basic. Writing the reaction as:
\[ \ce{H2O_{(l)} + H2O_{(l)} = H3O+_{(aq)} + OH-_{(aq)}} \tag{72}\]
highlights the fact that H3O+ is the conjugate acid of H2O, and OH- is the conjugate base of H2O. For convenience we may write,
\[ \ce{H2O_{(l)} = H+_{(aq)} + OH-_{(aq)}} \tag{73}\]
with the implicit understanding that H+ is present as H3O+. The equilibrium constant for water dissociation is given the special symbol Kw. The value is 1.0 x 10-14 at 25°C. The equilibrium constant is a function of temperature (as it is for all equilibria). For pure water at equilibrium the concentrations of H+ and OH- must be equal, as per reaction (73). Hence the equilibrium concentrations of H+ and OH- are both 1 x 10-7 M at 25°C. This is the point of neutrality.
Incidentally, pure sulfuric acid is also only weakly dissociated:
\[ \ce{2H2SO4_{(l)} = H3SO4+_{(aq)} + HSO4-_{(aq)}} \tag{74}\]
though more so than water; K = 2.7 x 10-4 at 25°C. (Other reactions also occur, making H2SO4 a more complex system.)
In aqueous solution, however, it fully (virtually) dissociates into:
\[ \ce{H2SO4_{(aq)} = H+_{(aq)} + HSO4-_{(aq)}} \tag{75}\]
Bisulfate is a weak acid:
\[ \ce{HSO4-_{(aq)} = H+_{(aq)} + SO4^2-_{(aq)}} \tag{76}\]
Ka2 = 0.012 at 25°C.
While Ka2 is <1, it is not extremely small.
Note: For convenience we often write sulfuric acid in solution as simply 2H+ + SO42-. Strictly speaking this is incorrect, but it is very common. It is understood that this is convenient shorthand for a system that includes H+, HSO4- and SO42-.
2.5 Other Types of Acids and Bases.
Even within the proton/hydroxide category there are a number of different types of acids and bases. We have already noted the strong and weak Lowry-Brønsted acids/bases.
1. Metal cations. Metal ions in solution exist as aquo complexes, as noted previously. Equilibria of the type,
\[ \ce{[M(H2O)_n]^{+z} = [M(H2O)_{n-1}OH]^{+(z-1)} + H+} \tag{77}\]
\[ \ce{[M(H2O)_{n-1}OH]^{+(z-1)} = [M(H2O)_{n-2}(OH)_2]^{+(z-1)} + H+} \tag{78}\]
may occur, as well as other types such as formation of hydroxo-bridged ions and oxo ions. These aquo complexes are all weak acids. Although in some instances they approach moderate strength. The first reaction is called the first ionization, and so on. In the case of metal ions this kind of acid-base behaviour is called hydrolysis. In principle this can continue successively. However, two things can start to occur as well. One is that the complexes can start to join together, e.g.
\[ \ce{[Fe(H2O)_6]^{3+} = [Fe(H2O)_5OH]^{2+} + H+} \quad K = 8.91 \times 10^{-4} \tag{79}\]
\[ \ce{[Fe(H2O)_5OH]^{2+} = [Fe(H2O)_4(OH)_2]^+ + H+} \quad K = 5.50 \times 10^{-4} \tag{80}\]
\[ \ce{2[Fe(H2O)_6]^{3+} = 2H+} \quad K = 0.00123 \tag{81}\]
The other is that oxide/hydroxide precipitates can start to form, e.g.
\[ \ce{[Fe(H2O)_6]^{3+} = Fe(OH)_3_{(s)} + 3H2O + 3H+} \tag{82}\]
\[ \ce{[Fe(H2O)_6]^{3+} = Fe(O)OH_{(s)} + 4H2O + 3H+} \tag{83}\]
\[ \ce{[Fe(H2O)_6]^{3+} = 1/2Fe2O3_{(s)} + 9/2H2O + 3H+} \tag{84}\]
depending on temperature. Other intermediate steps such as the acid dissociations shown above are also involved; these are just the overall reactions. Clearly the acid-base behaviour of ferric ion is complicated, and this kind of complexity is characteristic of many metal ions. A partial list of Ka values for the first dissociation i.e., reaction (77), of some aquo complexes in water is shown below. Note the huge range in values. Roughly speaking, the more highly charged cations are stronger acids.
| Table 2.2 - Some Ka values for the first dissociation of aquo complexes (on the molar; M scale). | |||||
|---|---|---|---|---|---|
| Main group | Transition metals | Lanthanides | |||
| Metal ion | Ka | Metal ion | Ka | Metal ion | Ka |
| Na+ | 2.0 x 10-15 | Ag+ | 1.3 x 10-12 | Ce+3 | 5.0 x 10-10 |
| Mg+2 | 4.0 x 10-12 | Zn+2 | 3.2 x 10-10 | Ce+4 | ~1 |
| Ba+2 | 5.0 x 10-14 | Pd+2 | 0.040 | Gd+3 | 2.5 x 10-9 |
| Al+3 | 1.0 x 10-5 | Fe+3 | 0.010 | ||
| Tl+1 | 5.0 x 10-14 | ||||
| Tl+3 | ~0.1 | ||||
2. Metal Oxides/Hydroxides. Metal oxides and hydroxides can exhibit amphoteric behaviour, i.e. acts as both weak acids and bases. This can be illustrated by some examples:
\[ \ce{Al(OH)_3_{(s)} + OH-_{(aq)} = [Al(OH)_4]^-_{(aq)}} \tag{85}\]
Here Al(OH)3 is acting as an acid towards OH- (quite in the Lewis sense, in fact). But we can also write reactions with acid:
\[ \ce{Al(OH)_3_{(s)} + H+_{(aq)} = Al(OH)_2^+_{(aq)} + H2O_{(l)}} \tag{86}\]
\[ \ce{Al(OH)_3_{(s)} + 3H+_{(aq)} = Al^{3+}_{(aq)} + 3H2O_{(l)}} \tag{87}\]
Now Al(OH)3 is acting as a base. The waters of coordination on Al+3 have been omitted for convenience. Oxides can also be amphoteric:
\[ \ce{ZnO_{(s)} + H2O_{(l)} = Zn^{2+}_{(aq)} + 2OH-_{(aq)}} \tag{88}\]
\[ \ce{ZnO_{(s)} + H2O_{(l)} = Zn(O)OH^-_{(aq)} + H^+_{(aq)}} \tag{89}\]
2.6 The pH scale
Acid and base concentrations span a huge range, from on the order of 10 M to <10-14 M. It is convenient to express acidity in terms of the logarithm of the activity of H+. This is called the pH:
\[ \text{pH} = -\log_{10} a_{H^+} \tag{90}\]
Again this is often approximated to -log10 [H+], and this will be generally used in this course. For pure water, or any solution where solution [OH-] = [H+] = 10-7 M, the pH is -log[H+] = 7.00. This is neutral pH. Since,
\[ K_w = \ce{[H^+][OH^-]} = 1.0 \times 10^{-14} \tag{91}\]
a pH of <7 implies that [H+] > [OH-], and the solution is then said to be acidic. Conversely, if pH > 7, then [OH-] > [H+] and the solution is basic.
In general “p” preceding any quantity indicates that -log10 of that quantity is meant. Hence,
\[ \text{pOH} = -\log_{10} a_{OH^-} = -\log \ce{[OH^-]} \tag{92}\]
In log form,
\[ \text{p}K_w = \text{pH} + \text{pOH} = 14 \ (\text{at 25°C}) \tag{93}\]
Then,
\[ \text{pOH} = 14 - \text{pH} \tag{94}\]
Acid-base conditions, principles and operating practice are very often presented in terms of pH, so it is very important that this be understood. Note some of the features of pH and activity/concentration:
• As pH decreases, the acidity (concentration of H+) increases.
• As pH increases the concentration of OH- increases.
• The pH of an aqueous solution becomes negative when aH+ > 1. This may occur in very concentrated solutions.
• It is often stated that the normal pH range for aqueous solutions is 0 < pH < 14. However, concentrated solutions of strong acids or bases can readily exceed this range.
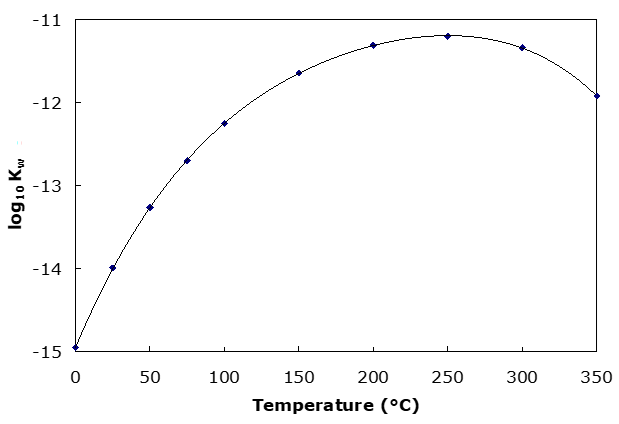
The value of the water dissociation constant, Kw, has a significant temperature dependence. This is illustrated in the figure below. In the figure the log Kw is plotted. Note that Kw rises then falls again.
2.7 Activity and concentration.
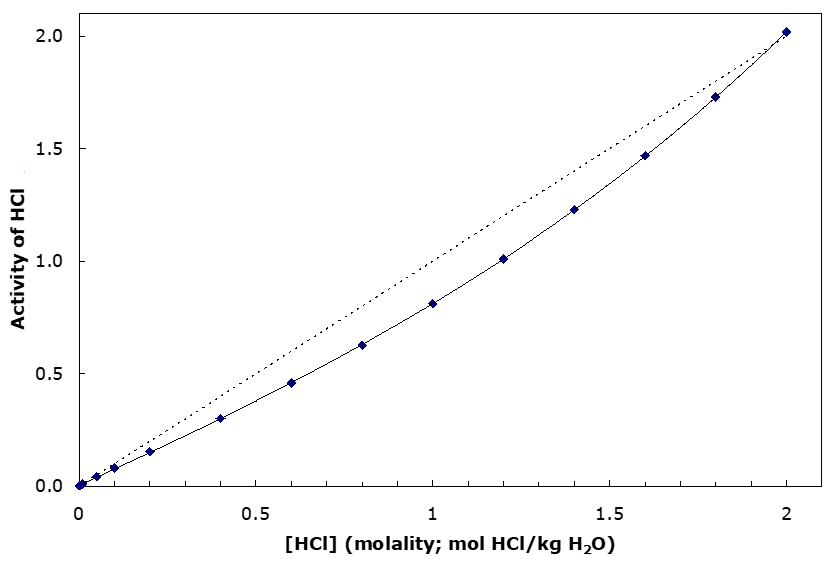
As has been noted before, activity is a kind of effective concentration. Water solutions are highly non-ideal. As an illustration of this point, the activity of HCl in water at 25°C as a function of concentration (in molality) is shown in Figure 2.2. Of course, in water HCl virtually fully dissociates into H3O+ and Cl-. But, since both species are present together in equal concentrations, we can’t measure their separate effects (at least not without extreme difficulty). All we can measure is their combined effects. (And we can’t generate a solution where there is an excess of H+ over Cl-, or vice versa; the energies involved in effecting bulk charge separation would be enormous. If we could we might be able to measure the effects of one ion over the other. But such a solution is physically impossible.) The graph quantitatively shows that there is a fairly significant error associated with assuming ideal behaviour, except at quite low concentration. Note too that past 2 m concentration the activity begins to exceed the concentration. Normal concentrated HCl has a concentration of about 12 M (mol/L); 15.9 m (molal); the density of concentrated HCl is 1.19 g/mL. Also, departure from ideality becomes more extensive as the charge of the ions increases.
2.8 Equilibrium Calculations for Acids and Bases.
A table of acid dissociation constants for a number of weak acids is provided in Table 2.3. Data are provided as pKa values:
\[ K_a = 10^{-\text{p}K_a} \tag{95}\]
1. Relating Ka and Kb for Conjugate Acids and Bases.
For the general acid dissociation,
\[ \ce{HA = H^+ + A^-} \tag{96}\]
we have,
\[ K_a = \frac{\ce{[H^+][A^-]}}{\ce{[HA]}} \tag{97}\]
But, A- is also a base:
\[ \ce{A^- + H2O = HA + OH^-} \tag{98}\]
\[ K_b = \frac{\ce{[HA][OH^-]}}{\ce{[A^-]}} \tag{99}\]
| Table 2.3 - Acid dissociation constants for a number of weak acids at 25°C | |||||
|---|---|---|---|---|---|
| Name | Acid1 | >pKa1 | pKa2 | pKa3 | pKa4 |
| Acetic acid | CH3CO2H | 4.76 | |||
| Ammonium | NH4+ | 9.24 | |||
| Arsenic acid | H3AsO4 | 2.24 | 6.96 | 11.50 | |
| Arsenious acid 2 | As(OH)3 | 9.29 | |||
| Benzoic acid | C6H5CO2H | 4.20 | |||
| Boric acid | B(OH)3 | 9.24 | 12.7 | 13.8 | |
| Carbonic acid | CO2 aq (“H2CO3”), HCO3- | 6.35 | 10.33 | ||
| Chromic acid | H2CrO4 | -0.2 | 6.51 | ||
| Chlorous acid | HClO2 | 1.95 | |||
| Hypochlorous acid | HOCl | 7.53 | |||
| Citric acid | HO2CCH2C(CO2H)(OH)CH2CO2H | 3.13 | 4.76 | 6.40 | |
| Diethylamine-H+ | (C2H5)2NH2+ | 10.93 | |||
| Ethylamine-H+ | C2H5NH3+ | 10.64 | |||
| Triethlamine-H+ | (C2H5)3NH+ | 10.72 | |||
| Ethylenediamine-(H+)2 | +H3NCH2CH2NH3+ | 6.85 | 9.93 | ||
| EDTA-(H+)2 3 | [(HO2CCH2)2NHCH2CH2NH(CH2CO2H)2]2+ | 0.0 | 1.5 | 2.0 | 2.68 |
| Formic acid | HCO2H | 3.75 | |||
| Guanidine | (H2N)2C=NH2+ | 13.54 | |||
| Hydrogen cyanide | HCN aq | 9.21 | |||
| Hydrofluoric acid | HF aq | 3.17 | |||
| Hydrogen peroxide | H2O2 aq | 11.65 | |||
| Hydrogen sulfide | H2S aq | 6.99 | 18.5 4 | ||
| Phenol | C6H5OH | 9.98 | |||
| Iodic acid | HIO3 | 0.77 | |||
| Nitrous acid | HNO2 | 3.15 | |||
| Oxalic acid | HOC(=O)C(=O)OH | 1.25 | 4.27 | ||
| Phosphoric acid | H3PO4 | 2.15 | 7.20 | 12.15 | |
| Phosphorous acid | H3PO3 | 1.5 | 6.79 | ||
| Pyridine | C5H5NH+ | 5.23 | |||
| Silicic | H2SiO3 | 9.91 | 11.90 | ||
| Selenic acid | H2SeO4 | strong | 1.92 | ||
| Selenious acid | H2SeO3 | 2.46 | 7.31 | ||
| Sulfuric | H2SO4 | strong | 1.99 | ||
| Sulfurous | SO2 aq + H2O = HSO3- + H+, HSO3- | 1.91 | 7.18 | ||
| Telluric acid | Te(OH)6 | 7.68 | 11.29 | ||
| Tellurous acid | H2TeO3 | 2.48 | 7.70 | ||
| 1. The acidic protons are indicated with bold H. | |||||
| 2. Also, pKa5 = 6.11; pKa2 = 10.17. | |||||
| 3. Some dissociations are too weak to detect, e.g. pKa2, 3 of As(OH)3. | |||||
| 4. Often ~14 is quoted in the hydrometallurgical context; this is probably too high in light of recent studies. | |||||
Since HA and A- differ by only on H+, it should be possible to related Ka and Kb. This can be done through Kw. We can rewrite equation (99):
\[ K_b = \frac{\ce{[HA][OH^-][H^+]}}{\ce{[A^-][H^+]}} \tag{100}\]
and bearing in mind that,
\[ K_w = \ce{[H^+][OH^-]} \tag{101}\]
\[ K_b = \frac{\ce{[HA]}}{\ce{[A^-][H^+]}} \quad K_w = \frac{K_w}{K_a} \tag{102}\]
Hence,
\[ K_a \cdot K_b = K_w \tag{103}\]
Note: This applies only for an acid and its conjugate base.
Note: It is a simple matter to relate pKa and pKb for conjugate acids and bases:
\[ -\log(K_a K_b) = -\log(10^{-14}) = -\log K_a + (-\log K_b) \tag{104}\]
\[ \text{p}K_a + \text{p}K_b = 14 \tag{105}\]
\[ \text{p}K_b = 14 - \text{p}K_a \tag{106}\]
Example 1
Calculate Kb for NH3 given that Ka for NH4+ is 5.6 x 10-10 (Table 2.3).
\[ K_b(\ce{NH3}) = \frac{K_w}{K_a(\ce{NH4^+})} = \frac{1 \times 10^{-14}}{5.75 \times 10^{-10}} = 1.74 \times 10^{-5} \tag{107}\]
This shows that if HA is a weak acid, then its conjugate base is a weak base. It also indicates that the stronger an acid, the weaker its conjugate base. This is illustrated in Table 2.4.
| Table 2.4 - Relationship between Ka and Kb for conjugate acids and bases. | |
|---|---|
| Ka | Kb |
| 1 x 10-14 | 1 |
| 1 x 10-10 | 1 x 10-4 |
| 1 x 10-7 | 1 x 10-7 |
| 1 x 10-4 | 1 x 10-10 |
| 1 | 1 x 10-14 |
Example 2
Calculate Kb for HS- and S2-. For H2S the conjugate acid is HS-, and for HS- it is S2-. Ka1 = 1.02 x 10-7 and Ka2 = 3.2 x 10-19 (Table 2.3). (Ka1 refers to the first acid dissociation constant, for H2S; Ka2 to the second, for HS-.)
\[ K_b(\ce{HS^-}) = \frac{K_w}{K_{a1}} = \frac{1 \times 10^{-14}}{1.02 \times 10^{-7}} = 9.8 \times 10^{-8} \tag{108}\]
\[ K_b(\ce{S^{2-}}) = \frac{K_w}{K_{a2}} = \frac{1 \times 10^{-14}}{3.2 \times 10^{-19}} = 3.1 \times 10^{4} \tag{109}\]
Sulfide is actually quite a strong base.
Note: that we cannot calculate Kb for S2- from Ka1 for H2S using equation (103); H2S and S2- are not conjugate acid-base pairs!
Example 3
Carbonic acid. As noted above, carbon dioxide in water exists as both CO2 and H2CO3. Ka1 can be taken to be due to both CO2 aq and H2CO3 together:
\[ \ce{CO2_{(aq)} + H2O_{(l)} = H^+_{(aq)} + HCO3^-_{(aq)}} \tag{110}\]
\[ \ce{CO2_{(aq)} + H2O_{(l)} = H2CO3_{(aq)}} \tag{111}\]
\[ \ce{H2CO3_{(aq)} = H^+_{(aq)} + HCO3^-_{(aq)}} \tag{112}\]
\[ K_{a1} = \frac{\ce{[H^+][HCO3^-]}}{\ce{[CO2] + [H2CO3]}} = 4.45 \times 10^{-7} \tag{113}\]
For practical purposes, we may treat aqueous carbon dioxide as either one of the forms CO2 aq or H2CO3 aq.
Note: carbon dioxide is a gas at ordinary temperatures and pressures, and is only weakly soluble in water,
\[ \ce{CO2_{(g)} = CO2_{(aq)}} \quad K = 0.034 \tag{114}\]
Hence Ka1 for CO2 gas, i.e.
\[ \ce{CO2_{(g)} + H2O_{(l)} = HCO3^-_{(aq)} + H^+_{(aq)}} \tag{115}\]
will not be the same as for reaction (111):
\[ \ce{CO2_{(aq)} + H2O_{(l)} = H^+_{(aq)} + HCO3^-_{(aq)}} \quad K_{a1} = 4.45 \times 10^{-7} \tag{116}\]
\[ \ce{CO2_{(g)} = CO2_{(aq)}} \quad K = 0.034 \tag{117}\]
\[ \ce{CO2_{(g)} + H2O_{(l)} = H^+_{(aq)} + HCO3^-_{(aq)}} \tag{118}\]
\[ K_{a1}(\ce{CO2_{(g)}}) = 0.034 \times 4.45 \times 10^{-7} = 1.5 \times 10^{-8} \tag{119}\]
Therefore it is very important that the phases of the acids/bases be indicated.
2. Calculation of Equilibrium Concentrations.
For simple monoprotic acids (one ionizable proton, also called monobasic acids) and bases that exhibit only one dissociation step, the calculation of equilibrium compositions is straightforward as the following examples illustrate:
Cyanide is a very important complexing agent in gold extraction. It is a weak base with pKa = 9.21 (Table 2.3). Calculate the equilibrium composition of a 1 g/L NaCN solution and calculate the pH.
\[ \frac{1\ \text{g/L NaCN}}{49.008\ \text{g/mol}} = 0.0204\ \text{M} \tag{120}\]
The reaction we need is,
\[ \ce{CN^-_{(aq)} + H2O_{(l)} = HCN_{(aq)} + OH^-_{(aq)}} \tag{121}\]
since it is cyanide ion that is mentioned (as NaCN, which is soluble and fully dissociates in ions; it is important that the solubility rules for salts be known). Hence we need Kb:
\[ \text{p}K_b = 14 - \text{p}K_a = 14 - 9.21 = 4.79 \tag{122}\]
\[ K_b = 10^{-4.79} = 1.62 \times 10^{-5} \tag{123}\]
The initial and equilibrium concentrations according to reaction (121) can be set up. Initially we have 0.0204 M CN-. Some of it dissociates. Let this be x.
\[ \ce{CN^-_{(aq)} + H2O_{(l)} = HCN_{(aq)} + OH^-_{(aq)}} \tag{124}\]
Initial: 0.0204
Change: x
\[ K_b = \frac{\ce{[HCN][OH^-]}}{\ce{[CN^-]}} = \frac{x^2}{0.0204 - x} \tag{125}\]
This resolves into a simple quadratic expression:
\[ x^2 + 1.62 \times 10^{-5}x - 0.0204K_b = 0 \tag{126}\]
\[ x = 5.668 \times 10^{-4}\ \text{M} = \ce{[OH^-]} = \ce{[HCN]} \tag{128}\]
At equilibrium then, [CN-] = 0.0198 M and [HCN] = [OH-] = 5.67 x 10-4 M. The pH is given by:
\[ \text{pH} = 14 - \text{pOH} = 14 - (-\log(5.668 \times 10^{-4})) = 10.75 \tag{129}\]
Cyanide also is a very toxic substance. When it hydrolyzes to form HCN (because CN- is a weak base), there is also a danger of toxic HCN gas escaping from the solution, which would pose a serious danger to workers. Care must be taken to keep the pH high enough so that [HCN] in solution is very low. This is done by keeping the pH above ~11.2. How much NaOH in g/L must be added to the above solution to achieve this pH?
We may again use the same dissociation reaction since all species involved are represented by this equilibrium. Now we know what the [OH-] must be:
\[ \text{pH} = 11.2;\ \text{pOH} = 14 - 11.2 = 2.8 \tag{130}\]
\[ \ce{[OH^-]} = 10^{-2.8} = 1.585 \times 10^{-3}\ \text{M} \tag{131}\]
There are two contributions to the OH- concentration: reaction (132) and added NaOH. Both must be factored in. Let the equilibrium [HCN] = x:
\[ \ce{CN^-_{(aq)} + H2O_{(l)} = HCN_{(aq)} + OH^-_{(aq)}} \tag{132}\]
table
Initial 0.0204
Change: x
\[ K_b = \frac{1.585 \times 10^{-3} \cdot x}{0.0204 - x} = 1.62 \times 10^{-5} \tag{133}\]
\[ x = 2.064 \times 10^{-4}\ \text{M} = \ce{[HCN]} \tag{134}\]
This is also the concentration of OH- that arises from CN- dissociation. The balance comes from added NaOH:
\[ \ce{Added\ NaOH} = 1.585 \times 10^{-3} - 2.064 \times 10^{-4} = 1.379 \times 10^{-3}\ \text{M} \tag{135}\]
\[ = 1.379 \times 10^{-3}\ \text{M} \tag{136}\]
The equilibrium constant for the reaction below is 7.1, i.e.
\[\ce{HCN(g) -> HCN(aq)}\tag{137}\]
\[\frac{[\ce{HCN}]}{P_{\ce{HCN}}} = 7.1\tag{138}\]
Assuming the solution concentration does not change, what is the maximum concentration of HCN in air over this solution in ppm? (1 ppm for gases = 1 mol/106 mol, or 1 mL/m3; The LC50 for HCN is about 3400 ppm for humans for a 1 minute exposure, but can be on the order of only 100 ppm for lengthier exposures. LC50 refers to the concentration of a gas in air that is lethal in 50% of the cases.)
\[P_{\ce{HCN}}\;(\text{atm})= \frac{[\ce{HCN}]}{7.1}= \frac{2.064 \times 10^{-4}}{7.1}= 2.907 \times 10^{-5}\ \text{atm} \tag{139} \]
Assuming that atmospheric pressure is 1 atm, and using the ideal gas law, PV = nRT,
\[\frac{P_{\ce{HCN}}}{1}= \frac{n_{\ce{HCN}}}{n_{\text{total}}}= 2.91 \times 10^{-5}\tag{140}\]
This is the fraction of the total gas that is HCN. In ppm it is:
\[2.91 \times 10^{-5} \times 1 \times 10^{6}= 29\ \text{ppm}\tag{141}\]
This is below the toxic limit, assuming that no pooling occurs. (HCN gas is heavier than air.) Diffusion and air currents would dilute the concentration farther from the surface.
Calculate the fraction of the total cyanide in % that is present as CN- as a function of pH. Plot this.
Here we do not know the actual starting or total [CN-]. This is easier than it looks. For the acid dissociation,
\[\ce{HCN(aq) <=> H+ (aq) + CN- (aq)}\tag{142}\]
\[K_\mathrm{a}= \frac{[\ce{H+}][\ce{CN-}]}{[\ce{HCN}]}= 10^{-pK_\mathrm{a}}= 10^{-9.21}= 6.17 \times 10^{-10} \tag{143}\]
\[[\ce{CN-}]= \frac{K_\mathrm{a}[\ce{HCN}]}{[\ce{H+}]}= \frac{K_\mathrm{a}[\ce{HCN}]}{10^{-pH}}\tag{144}\]
Fraction of the total present as HCN is:
\[\frac{[\ce{CN-}]}{[\ce{HCN}] + [\ce{CN-}]}=\frac{\dfrac{K_\mathrm{a}[\ce{HCN}]}{10^{-pH}}}{[\ce{HCN}] +\dfrac{K_\mathrm{a}[\ce{HCN}]}{10^{-pH}}}\tag{145}\]
\[= \frac{K_\mathrm{a} / 10^{-pH}}{K_\mathrm{a} / 10^{-pH} + 1}\tag{146}\]
A table can be set up with the results of the calculations over a range of pH.
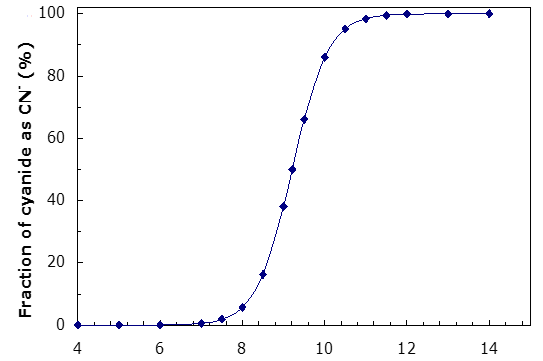
| Table 2.5 - Fraction of total cyanide present as CN- as a function of pH at 25°C | |
|---|---|
| pH | Fraction as HCN % |
| 4 | 99.999 |
| 5 | 99.994 |
| 6 | 99.938 |
| 7 | 99.387 |
| 8 | 94.192 |
| 9 | 61.858 |
| 9.21 | 50.000 |
| 10 | 13.955 |
| 11 | 1.596 |
| 12 | 0.162 |
| 13 | 0.0162 |
| 14 | 0.00162 |
Note: At at pH = 9.21 = pKa the fraction of HCN is 50% of the total. This is always the case for a monoprotic acid. At pH = pKa, half is present as the acid and half as the conjugate base. For polyprotic acids this is also very close to true so long as the pKa values are sufficiently separated.
3. Neutralization.
The term has two meanings. One is the addition of just enough base (or acid) to react with all of the acid (or base) present in a solution or mixture. The second is adding base to acid, or vice versa, to adjust the pH to some specified value. In this case either a deficiency or an excess is added relative to the stoichiommetric requirement. If this is meant the final solution pH must be specified.
1. Neutralization of a strong base or a strong acid is straightforward. The conjugate acid or base, respectively is extremely weak and does not affect the final pH.
How much NaOH kg/h is needed to neutralize 100 m3/h of 1 M HCl and what will the pH be for the final solution?
\[\ce{NaOH(s) + H+ (aq) + Cl- (aq) -> H2O(l) + Na+ (aq) + Cl- (aq)} \tag{147}\]
The acid and base react in a 1:1 mol ratio.
\[
100\ \frac{\text{m}^3}{\text{h}}
\times 1000\ \frac{\text{L}}{\text{m}^3}
\times \frac{1\ \text{mol HCl}}{\text{L}}
\times \frac{1\ \text{mol NaOH}}{\text{mol HCl}}
\times \frac{40.00\ \text{g NaOH}}{\text{mol NaOH}}
\times 10^{-3}\ \frac{\text{kg}}{\text{g}}
= 4000\ \text{kg/h}
\tag{148}
\]
The final solution contains Na+ and Cl-. Each is an exceedingly weak acid and base, respectively. The only significant acid-base equilibrium at play is the water dissociation. Hence [H+] = [OH-] = 10-7 M; pH = 7.0.
2. How much lime in g/L is needed to neutralize a 1 M H2SO4 solution and estimate what the final pH will be? The neutralization product is CaSO4·2H2O (gypsum). It is slightly soluble (about 0.01 M).
The reaction required is:
\[
\ce{H2SO4(aq) + CaO(s) + H2O(l) -> CaSO4.2H2O(s)}
\tag{149}
\]
\[
\frac{1\ \text{mol H}_{2}\text{SO}_{4}}{\text{L}}
\times \frac{1\ \text{mol CaO}}{\text{mol H}_{2}\text{SO}_{4}}
\times \frac{56.079\ \text{g CaO}}{\text{mol CaO}}
= 56.079\ \text{g CaO/L}
\tag{150}
\]
The solution in the end contains a small concentration of Ca+2 and SO42-, about 0.01 M of each. Calcium ion is a very weak acid and sulfate is a very weak base. According to Table 1, we might expect Ka for [Ca(H2O)6]+2 to be <4 x 10-12 (= Ka for Mg+2(aq)), but > 5 x 10-14 (= Ka for Ba+2(aq)). A value on the order of 10-12 is a reasonable guess. Kb for sulfate is given by:
\[
K_\mathrm{b}(\ce{SO4^{2-}})
= \frac{K_\mathrm{w}}{K_\mathrm{a}(\ce{HSO4-})}
= \frac{1.0 \times 10^{-14}}{0.0102}
= 9.8 \times 10^{-13}
\tag{151}
\]
(See Table 2.3 for pKa of HSO4-.) Sulfate is a very weak base. The pH due to sulfate dissociation would be given by:
\[
\ce{SO4^{2-} + H2O <=> HSO4- + OH-}
\tag{152}
\]
Initial: 0.01
Change: x
\[
\frac{x^{2}}{0.01 - x}
\approx
\frac{x^{2}}{0.01}
= 9.8 \times 10^{-13}
\qquad
\text{since } x \ll 0.01,\; K_\mathrm{b} \ll 1
\tag{153}
\]
\[
x = 9.5 \times 10^{-8}\ \text{M}
= [\ce{OH-}]
\tag{154}
\]
This is comparable to [OH-] in neutral water. Next, Ca+2(aq) is also a weak acid, with a Ka numerically very similar to the Kb for sulfate. Again then the extent of dissociation is expected to be very small, and comparable to neutral water. On balance then we would expect a final pH to be quite close to 7.
3. Neutralization of a weak acid by a strong base, or a weak base by a strong acid is a bit more involved. Taking the first case, where the weak acid is depicted as HA,
\[
\ce{HA_{(aq)} + OH^-_{(aq)} -> A^-_{(aq)} + H2O_{(l)}}
\tag{155}
\]
we have all of the HA (both undissociated HA plus dissociated as H+ and A-) converted to A-. This is what stoichiommetric neutralization means. But, A- is a weak base, and so once the neutralization reaction is done A- will partially dissociate, as governed by its Kb, to form a small concentration of HA and OH-:
\[
\ce{A^-_{(aq)} + H2O_{(l)} <=> HA_{(aq)} + OH^-_{(aq)}}
\tag{156}
\]
The final solution then will be somewhat basic and not at neutral pH. The final pH may be readily calculated. This type of situation is exactly like that of calculating the composition of a solution of a weak base, or of a weak acid if neutralizing a weak base with strong acid.
Consider the example of a solution of 0.1 M HF to be neutralized with 2 M NaOH. Hydrofluoric acid is a decomposition production of fluorosilicic acid (H2SiF6), used in lead processing. How much NaOH solution is needed per litre of HF solution and calculate the final pH? The neutralization reaction is:
\[
\ce{HF_{(aq)} + OH^-_{(aq)} -> H2O_{(l)} + F^-_{(aq)}}
\tag{157}
\]
The Ka for HF is 10-3.17 = 6.76 x 10-4 (Table 2.3). Some of the hydrofluoric acid is present as H+ and F-, and most as HF. The sum [H+] + [HF] = 0.1 M. All the H+, either free or bound to HF must be neutralized. Mathematically we can treat it as if it is all HF.
Upon addition of NaOH solution, we will form a solution of F-, a weak base, at a lower concentration than the original 0.1 M, due to dilution. Hence we need to know the volume of 1 M NaOH added. After that the problem is on familiar ground.
\[
\frac{0.1 \text{ mol HF}}{L HF{aq}} \times
\frac{1 \text{ mol OH}^-}{\text{ mol HF}} \times
\frac{1 \text{ L NaOH}_{aq}}{2 \text{ mol OH}^-}
= \frac{0.05 \text{ L of 2 M NaOH}}{L HF{aq}}
=0.05\ L\ NaOH{aq}/L\ HF{aq}\tag{158}
\]
The [F-] after neutralization is 0.1 mol/1.05 L = 0.09524 M (0.05 L 2 M NaOH added to 1 L of 0.1 M HF). Now the pH can be calculated. At equilibrium we have:
\[
\ce{F^-_{(aq)} + H2O_{(l)} <=> HF_{(aq)} + OH^-_{(aq)}}
\tag{159}
\]
Initial: 0.09524
Final 0.09524 - x x x
\[
\frac{x^{2}}{0.09524 - x}
= K_\mathrm{b}
= \frac{1.0 \times 10^{-14}}{6.76 \times 10^{-4}}
= 1.48 \times 10^{-11}
\tag{160}
\]
Solving this gives x = 1.19 x 10-6 M = [OH-] = [HF]. Then,
\[
\mathrm{pOH}
= -\log(1.19 \times 10^{-6})
= 5.92
\tag{161}
\]
\[
\mathrm{pH}
= 14 - \mathrm{pOH}
= 8.08
\tag{162}
\]
3. Buffers.
There are different kinds of buffers, including pH buffers, metal ion buffers and potential buffers. A pH buffer is a solution or a mixture (e.g. of a solid and a solution) that resists change in pH. Specifically, if a not too large amount of an acid is added the pH drops only a little, and if some of a strong base is added, the pH rises only a little. The changes are markedly less than would be the case if a buffer system were not in place.
There are also different types of pH buffers. First we will deal with those derived from an acid and its conjugate base (which is why the conjugate acid-base idea is so important). Next we will deal with buffers comprised of salts of weak acids and weak bases. Finally, solid metal oxides, or hydroxides plus a soluble salt of the same metal ion also act as buffers. These will be dealt with in the section on the solubility product (Ksp), which has to do with sparingly soluble salts.
Two conditions are needed for a solution to act as a buffer in the manner of the first case:
• An acid and its conjugated base must be present in the solution.
• Both must be present at similar and substantial concentrations.
Consider a solution as above, with similar and substantial concentrations of HA and A-. If a small amount of NaOH is added, it will react according to:
\[
\ce{HA + OH^- -> H2O + A^-}
\tag{163}
\]
This reaction is just the opposite of the base dissociation reaction for A-:
\[
\ce{H2O + A^- <=> HA + OH^-}
\tag{164}
\]
Since most weak bases have Kb << 1, the equilibrium constant for reaction (163) will be large, i.e. 1/Kb. Therefore, the strong base OH- is substantially converted into the weak base A-. And since A- is a weak base, it dissociates to only a small extent, so the pH rises only a little, and much less than it would have in the absence of the buffer.
Similarly if a small amount of a strong acid is added it reacts almost completely with the base A-:
\[
\ce{A^-_{(aq)} + H^+_{(aq)} -> HA_{(aq)}}
\qquad
K = \frac{1}{K_\mathrm{a}}
\tag{165}
\]
Again, in most cases Ka << 1 so K >> 1 and the strong acid H+ is almost completely converted to the weak acid HA. This dissociates only a little, so the pH drops to only a small extent. This is how a buffer of this type works. (If one or both of the acid and conjugate base are only weakly soluble, this mixture too may function as a buffer, and by the same principles.)
The effects are readily quantified by means of Ka. We can express Ka in logarithmic terms and rearrange as follows:
\[
\ce{HA_{(aq)} <=> H^+_{(aq)} + A^-_{(aq)}}
\tag{166}
\]
\[
\log K_\mathrm{a}
= \log\!\left(\frac{[H^+][A^-]}{[HA]}\right)
\tag{167}
\]
\[
\log K_\mathrm{a}
= \log[H^+] + \log\!\left(\frac{[A^-]}{[HA]}\right)
\tag{168}
\]
\[
-\log K_\mathrm{a}
= -\log[H^+] - \log\!\left(\frac{[A^-]}{[HA]}\right)
\tag{169}
\]
\[
\mathrm{p}K_\mathrm{a}
= \mathrm{pH} - \log\!\left(\frac{[A^-]}{[HA]}\right)
\tag{170}
\]
\[
\mathrm{pH}
= \mathrm{p}K_\mathrm{a} + \log\!\left(\frac{[A^-]}{[HA]}\right)
\tag{171}
\]
Alternatively for the base B and acid BH+, the quotient would be [B]/[BH+]. This will allow us to calculate the pH of a solution so long as we know [HA] and [A-].
1. Example calculation.
Reactions involving ammonia in hydrometallurgy often require buffering. Calculate the pH of a solution containing 1 M each of ammonia and ammonium sulfate.
Ammonium sulfate is (NH4)2SO4, and sulfates and ammonium salts are generally soluble in water. Hence 1 M (NH4)2SO4 = 2M NH4+. The relevant reaction is:
\[
\ce{NH4^+_{(aq)} <=> NH3_{(aq)} + H^+_{(aq)}}
\tag{172}
\]
Initial 2 1
Final 2-x 1+x x
If x M of NH4+ dissociates then x M additional NH3 forms, and x M H+.
\[
K_\mathrm{a}
= \frac{x(1 + x)}{2 - x}
= 10^{-9.24}
= 5.75 \times 10^{-10}
\tag{173}
\]
This resolves into a quadratic and solving gives x = 1.15 x 10-9. Obviously this is extremely small; x << 1 or 2. The implication is that [NH3] and [NH4+] at equilibrium are almost identical to the initial values. Why? Substantial concentrations of both are present. NH4+ is a weak acid. It will dissociate very little. NH3 is a weak base. It too will dissociate very little. And the presence of one suppresses dissociation of the other, as per Le Chatelier’s principle. Hence there will be little change in the concentrations. In general, we can make this assumption for a buffer solution:
\[
[HA]_{\text{initial}} = [HA]_{\text{final}}
\qquad
\text{and}
\qquad
[A^-]_{\text{initial}} = [A^-]_{\text{final}}
\tag{174}
\]
so long as both are present at substantial and similar concentrations. The only other important caveat is that one of Ka or Kb cannot be very small (comparable to or less than Kw). If that is the case, then reaction with strong base or acid, respectively, may be incomplete and equation [174] may not pertain.
Continuing with the ammonia/ammonium example, pKa for NH4+ = 9.24
\[
\mathrm{pH}
= 9.24 + \log\!\left(\frac{1}{2}\right)
= 8.94
\tag{175}
\]
2. The Buffer Point of an Acid.
Consider equation (171) again. When [A-] = [HA], or equivalently [B] = [BH+], then,
\[
\mathrm{pH}
= \mathrm{p}K_\mathrm{a} + \log(1)
= \mathrm{p}K_\mathrm{a}
\tag{176}
\]
At this point we have equal concentrations of the acid and its conjugate base. This is called the buffer point of an acid. The smaller Ka is, i.e. the larger pKa, the higher is the buffer point. It buffers at a higher pH. Thus the buffer point for HF is 3.17 (Table 2.3). For HCN it is 9.21, etc.
3. Sulfate/Bisulfate Buffering.
This is very important in many hydrometallurgical processes. The majority of hydrometallurgical processes employ acidic sulfate medium. In leaching, for instance, a substantial concentration of sulfate (SO42-) will build up in solution, e.g. from reaction with metal oxides:
\[
\ce{MO_{(s)} + H2SO4_{(aq)} -> M^{2+}_{(aq)} + SO4^{2-}_{(aq)} + H2O_{(l)}}
\tag{177}
\]
If acid is added as H2SO4 we will have a solution containing H+, HSO4- and SO42-. The pH will not drop as far as it would in the absence of substantial sulfate concentration. The solution is buffered. This limits the ability to obtain a very low pH with sulfuric acid.
\[
\ce{H2SO4_{(aq)} -> H^+_{(aq)} + SO4^{2-}_{(aq)}}
\qquad
K \text{ is very large}
\tag{178}
\]
The additional H+ will partially react with SO42- and form HSO4-:
\[
\ce{SO4^{2-}_{(aq)} + H^+_{(aq)} <=> HSO4^-_{(aq)}}
\tag{179}
\]
Based on the preceding discussion, we might expect this reaction to go largely to completion. The Ka for HSO4- is 0.0102 (Table 2.3). What is K for reaction (92)?
\[
K
= \frac{1}{K_\mathrm{a}(\mathrm{HSO_4^-})}
= \frac{1}{0.0102}
= 98.0
\tag{180}
\]
This is not exceedingly large, and illustrates an earlier point that very small equilibrium constants for Ka or Kb may invalidate the assumption that initial and equilibrium concentrations of weak acid/weak base are almost equal. What are the equilibrium concentrations for a sulfate medium with 0.2 M sulfate and 0.2 M bisulfate? Consider the reaction:
\[
\ce{HSO4^-_{(aq)} <=> H^+_{(aq)} + SO4^{2-}_{(aq)}}
\tag{181}
\]
Initial 0.2 0.2 0.2 - x x 0.2 + x
\[
K_\mathrm{a}
= \frac{x\,(0.2 + x)}{\,0.2 - x\,}
\tag{182}
\]
Solving the quadratic gives x = 0.00929 M. This is 4.6% of 0.2 M, which is small, but not inconsequential. Having the equilibrium concentrations the pH can be found:
\[
\mathrm{pH}
= \mathrm{p}K_\mathrm{a} + \log[\,\mathrm{SO_4^{2-}}\,]
\tag{183}
\]
\[
\mathrm{pH}
= 1.99
+ \log\!\left(\frac{0.2 + 0.00929}{0.2 - 0.00929}\right)
\tag{184}
\]
pH = 2.03
Had we made the assumption that [HSO4-] = [SO42-] = 0.2, the pH would have been calculated to be 1.99, which is still quite close. But, it is clear now that our assumption that [HA]initial = [HA]final, and likewise the base concentration, is imperfect. If a base or acid is very weak, this will be the case and the method outlined here is best used to determine the equilibrium concentrations.
4. Effects of Concentration and pKa on Buffering.
It is helpful to see, quantitatively, the effects of buffering on pH. This section will explore this. Concentration of the acid and its conjugate base affect the buffer pH significantly, as equation (171) indicates. It is important that the buffer have similar concentrations of the both. If [HA] is high and [A-] is low, addition of a little strong base will react with HA to form A-, but the change in [A-] might be substantial, since there was little of it to begin with. The affect of the strong base OH- on the pH is attenuated, but not as much as if [A-] has been higher.
Similarly, if a little strong acid is added then we will use up a significant amount of the A-; there is not much of it to react with the added H+. The strong acid H+ might be converted to weak acid HA, but the change in [A-] is significant. The pH change will not be as great as it would have been in the absence of the buffer, but it will be greater than if [A-] had been higher. The same argument can be applied to a solution with low [HA] and high [A-]. Hence a buffer has the highest capacity to resist change in pH when [A-] = [HA]. It should be clear too that the higher the concentration of HA and A-, the greater the buffer’s capacity to absorb strong base or strong acid with little change in pH. These effects can be quantified.
First, NH3 is a stronger base than NH4+ is an acid; Kb >> Ka. Hence the chemical reaction involving NH3 and NH4+ is best written as:
\[
\ce{NH3_{(aq)} + H2O_{(l)} <=> NH4^+_{(aq)} + OH^-_{(aq)}}
\tag{185}
\]
Using the acid dissociation reaction will lead to errors and confusion. Note: This is an important point! Write the reaction in the direction that it is most likely to go. We will start with [NH3] + [NH4+] = 1 M and look at the buffering effect as a function of different relative amounts of the two. For differing ratios of r = [NH3]/[NH4+] the concentrations of each can be found:
\[
[\mathrm{NH_3}]
= 1 - [\mathrm{NH_4^+}]
\tag{186}
\]
\[
1-\frac{[\mathrm{NH_4^+}]}{[\mathrm{NH_4^+}]}
= r
\tag{187}
\]
Rearranging gives,
\[
[\mathrm{NH_4^+}]
= \frac{1}{1 + r}
\tag{188}
\]
We can calculate the pH using equation (171), either assuming that the initial and final concentrations are the same (that they change very little due to negligible dissociation), or by calculating the equilibrium concentrations based on Kb. To do the latter, let Co = [NH3]initial and Co’ = [NH4+]initial. Let x M of NH3 dissociate at equilibrium. (If we wrote the reaction the other way round, we might let x M of NH4+ dissociate, but since Kb > Ka, then x would have to be negative.)
\[
\ce{NH3_{(aq)} + H2O_{(l)} <=> NH4^+_{(aq)} + OH^-_{(aq)}}
\tag{189}
\]
Co - x Co’ + x x
\[
K_\mathrm{b}
= \frac{x\,(C_0' + x)}{C_0 - x}
= 1.738 \times 10^{-5}
\tag{190}
\]
Rearranging gives,
\[
x^{2}
+ (C_0' + K_\mathrm{b})\,x
- K_\mathrm{b}\,C_0
= 0
\tag{191}
\]
\[
x
= \frac{-(C_0' + K_\mathrm{b})
+ \sqrt{(C_0' + K_\mathrm{b})^{2} + 4K_\mathrm{b}C_0}}
{2}
\tag{192}
\]
The unknown, x, can be solved for various Co and Co’. A table of the results is shown below. First the pH was calculated assuming the equilibrium concentrations equal the initial values. On the right of the table the pH was calculated by taking into account changes in concentrations due to dissociation as per reaction (191). As can be seen the assumption is excellent below [NH3]/[NH4+] = 10, and poor when the ratio exceeds 100. (There is an asymmetry to this.)
| Table 2.6 - Calculated pH for solutions of ammonia and ammonium at different concentration ratios. Total [NH3] + [NH4+] = 1M. | |||||||
|---|---|---|---|---|---|---|---|
| [NH3] [NH4+] | Initial [NH3] |
Initial [NH4+] |
pH 1 | x | Equil. [NH3] |
Equil. [NH4+] |
pH 2 |
| 0.0001 | 1.00E-04 | 0.9999 | 5.24 | 1.738E-09 | 1.00E-04 | 1.00E+00 | 5.24 |
| 0.001 | 9.99E-04 | 0.999 | 6.24 | 1.738E-08 | 9.99E-04 | 9.99E-01 | 6.24 |
| 0.01 | 9.90E-03 | 0.990 | 7.24 | 1.738E-07 | 9.90E-03 | 9.90E-01 | 7.24 |
| 0.1 | 0.0909 | 0.909 | 8.24 | 1.738E-06 | 9.09E-02 | 9.09E-01 | 8.24 |
| 1 | 0.500 | 0.500 | 9.24 | 1.738E-05 | 5.00E-01 | 5.00E-01 | 9.24 |
| 10 | 0.909 | 0.0909 | 10.24 | 1.734E-04 | 9.09E-01 | 9.11E-02 | 10.24 |
| 100 | 0.990 | 9.90E-03 | 11.24 | 1.506E-03 | 9.89E-01 | 1.14E-02 | 11.18 |
| 1000 | 0.999 | 9.99E-04 | 12.24 | 3.689E-03 | 9.95E-01 | 4.69E-03 | 11.57 |
| 10000 | 0.9999 | 1.00E-04 | 13.24 | 4.110E-03 | 9.96E-01 | 4.21E-03 | 11.61 |
| 1. Assuming that initial concentrations = equilibrium concentrations. | |||||||
| 2. pH calculated after determining changes in concentrations of NH3 and NH4+. | |||||||
Next, we can see what effect adding some strong base, NaOH, has on the pH. To each mixture 0.2 g/L solid NaOH is added, or 0.005 mol/L. The OH- reacts with NH4+ almost quantitatively:
\[
\ce{NH4^+_{(aq)} + OH^-_{(aq)} <=> NH3_{(aq)} + H2O_{(l)}}
\tag{193}
\]
\[
K
= \frac{1}{K_\mathrm{b}}
= \frac{1}{1.738 \times 10^{-5}}
= 5.75 \times 10^{4}
\tag{194}
\]
Hence initial [NH4+] drops to Co’ - 0.005, and [NH3] increases to Co + 0.005. We again calculate the change in concentrations using equation (193). However, for the last two entries in Table 5, Co’ < added [OH-]. Here we take Co’ to be zero and an initial left over [OH-] to be 0.005 - Co’. Then we can use:
\[
\ce{NH3_{(aq)} + H2O_{(l)} <=> NH4^+_{(aq)} + OH^-_{(aq)}}
\tag{195}
\]
| Initial | Co | Co' | before adding NaOH | |
| + NaOH | 0.005 | added NaOH; Co' < 0.005 | ||
| New initial | Co + Co' | 0 | 0.005 - Co' | all NH4+ → NH3 |
| Equil. | Co + Co' - x | x | 0.005 - Co' + x | x NH3 dissociates |
\[
K_\mathrm{b}
= \frac{x\,(0.005 - C_0' + x)}{\,C_0 + C_0' - x\,}
\tag{196}
\]
\[
x^{2}
+ (0.005 - C_0' + K_\mathrm{b})\,x
- K_\mathrm{b}\,(C_0 + C_0')
= 0
\tag{197}
\]
This can be solved as usual and allows the composition to be calculated for the last two [NH3]/[NH4+] ratios. The results are tabulated below.
| Table 2.7 - Effect of adding solid 0.005 M NaOH to solutions from Table 5. Total [NH4 ] + [NH4+] = 1 M | |||||||
|---|---|---|---|---|---|---|---|
| Initial [NH4+] 1 | Initial [NH4+] 1 | Initial [NH4] [NH4+] |
Initial [OH-]1 | x | Equil. [NH4] | Equil. [NH4+] |
pH |
| 5.10E-03 | 0.9949 | 0.0051 | 0 | 1.74E-09 | 5.10E-03 | 0.9949 | 6.952 |
| 6.00E-03 | 0.9940 | 0.0060 | 0 | 1.74E-08 | 6.00E-03 | 0.9940 | 7.022 |
| 0.0149 | 0.9851 | 0.0151 | 0 | 1.74E-07 | 0.0149 | 0.9851 | 7.422 |
| 0.0959 | 0.9041 | 0.1061 | 0 | 1.74E-06 | 0.0959 | 0.9041 | 8.272 |
| 0.5050 | 0.4950 | 1.020 | 0 | 1.74E-05 | 0.5050 | 0.4950 | 9.252 |
| 0.9141 | 0.0859 | 10.64 | 0 | 1.73E-04 | 0.9139 | 0.0861 | 10.272 |
| 0.9951 | 4.90E-03 | 203.0 | 0 | 1.51E-03 | 0.9936 | 6.41E-03 | 11.433 |
| 1.0000 | 0 | n/a | 4.00E-03 | 2.62E-03 | 0.9974 | 2.62E-03 | 11.823 |
| 1.0000 | 0 | n/a | 4.90E-03 | 2.38E-03 | 0.9976 | 2.38E-03 | 11.863 |
| 1. “New initial” concentrations after adding NaOH and reaction with NH4+. | |||||||
| 2. pH calculated after determining changes in concentrations of NH3 and NH4+ by equation [96]. | |||||||
| 3. pH calculated after determining changes in concentrations of NH3 and NH4+ by equation [99]. | |||||||
The results from Table 2.6 and Table 2.7 are compared in Table 2.8.
| Table 2.8 - Comparison of pH of ammonia/ammonium solutions ([NH3] + [NH4+] = 1 M) before and after NaOH addition (0.005 M) | ||
|---|---|---|
| Initial [NH3] [NH4+] | pH 1 prior to NaOH addition |
pH 1 after NaOH addition |
| 0.0001 | 5.24 | 6.95 |
| 0.001 | 6.24 | 7.02 |
| 0.01 | 7.24 | 7.42 |
| 0.1 | 8.24 | 8.27 |
| 1 | 9.24 | 9.25 |
| 10 | 10.24 | 10.27 |
| 100 | 11.18 | 11.43 |
| 1000 | 11.57 | 11.82 |
| 10000 | 11.61 | 11.86 |
| 1. pH calculated after determining changes in concentrations of NH3 and NH4+. | ||
Note that the pH of a 0.005 M NaOH solution alone would be 14 + log 0.005 = 11.7. Adding this concentration of NaOH to the buffer with[NH3]/[NH4+] = 1 results in almost no change in pH. As the ratios depart from one, the pH changes more substantially (always higher; NaOH is a base). It is clear then that the further the ratio [NH3]/[NH4+] departs from 1, the less the capacity of the buffer to resist pH changes.
The same calculations can be done for HSO4-/SO42-. In this case Kb(SO42-) is very small (9.8 x 10-13). The effects of this on the buffer calculations can be checked. HSO4- is a stronger acid than SO42- is a base, so use the acid dissociation reaction as the basis for the calculations:
\[
\ce{HSO4^-_{(aq)} <=> H^+_{(aq)} + SO4^{2-}_{(aq)}}
\tag{198}
\]
Given a ratio [SO42-]/[HSO4-] and total [HSO4-] + [SO42-], the concentrations can be calculated in the same way as equation (190). To calculate the equilibrium compositions we start with:
\[
\ce{HSO4^-_{(aq)} <=> H^+_{(aq)} + SO4^{2-}_{(aq)}}
\tag{199}
\]
Initial Co Co’
Final Co - x x Co’ + x
\[
K_\mathrm{a}
= \frac{x\,(C_0' + x)}{C_0 - x}
= 0.0102
\tag{200}
\]
The calculations are done in the same way as the ammonia/ammonium case. The results is shown in Table 9. In this case serious errors arise at [SO42-]/[HSO4-] < 1. (Again we have an asymmetry, but opposite to that of the NH3/NH4+ case, due to Ka (HSO4-) > Kb (SO42-).) The calculations agree well when the ratio > 1.The disagreement is more serious for this system than for the sulfate/bisulfate system than for the ammonia/ammonium one. This highlights the effect of the very small Kb for SO42-. Note though that the buffer point pH (where [SO42-] = [HSO4-]) is only slightly in error when we assume the initial concentrations are equal to the equilibrium values.
| Table 2.9 - Calculated pH for solutions of sulfate and bisulfate at different concentration ratios. Total [SO42-] + [HSO4-] = 1 M. | |||||||
|---|---|---|---|---|---|---|---|
| [SO42-] / [HSO4-] | Initial [SO42-] | Initial [HSO4-] | pH1 | x | Equilibrium [SO42-] | Equilibrium [HSO4-] | pH2 |
| 0.001 | 9.99E-04 | 0.9990 | -1.01 | 9.56E-02 | 0.0966 | 0.9034 | 1.02 |
| 0.01 | 9.90E-03 | 0.9901 | -0.01 | 9.11E-02 | 0.1010 | 0.8990 | 1.04 |
| 0.1 | 0.0909 | 0.9091 | 0.99 | 5.83E-02 | 0.1492 | 0.8508 | 1.23 |
| 1 | 0.500 | 0.500 | 1.99 | 9.84E-03 | 0.5098 | 0.4902 | 2.01 |
| 10 | 0.9091 | 0.0909 | 2.99 | 1.01E-03 | 0.9101 | 0.0899 | 3.00 |
| 100 | 0.9901 | 9.90E-03 | 3.99 | 1.01E-04 | 0.9902 | 9.80E-03 | 3.99 |
| 1000 | 0.9990 | 9.99E-04 | 4.99 | 1.01E-05 | 0.9990 | 9.89E-04 | 4.99 |
| 1. Assuming that initial concentrations = equilibrium concentrations. | |||||||
| 2. pH calculated after determining changes in concentrations of SO42- and HSO4-. | |||||||
The effects of adding a small concentration of a strong acid (e.g. HCl gas) at 0.005 M to the above solutions are developed analogously to the treatment for the
ammonia/ammonium example. The reaction we expect is:
\[
\ce{SO4^{2-}_{(aq)} + H^+_{(aq)} <=> HSO4^-_{(aq)}}
\tag{201}
\]
which is the reverse of the acid dissociation.
\[
K
= \frac{1}{K_\mathrm{a}}
= \frac{1}{0.0102}
= 98.0
\tag{202}
\]
Although >1, it is not very large, and we would anticipate that the reaction will not proceed completely to the right. This will have implications for our buffer calculations. As previously, we assume starting compositions as if all the added H+ fully converts SO42- to HSO4-. Then we allow for dissociation and calculate equilibrium concentrations, as per equation (200). Again, for one solution the added [H+] exceeds the initial SO42-. In that case, initially we will have some left over H+. We can set up this situation as follows, analogously to the ammonia/ammonium case:
\[
\ce{HSO4^-_{(aq)} <=> SO4^{2-}_{(aq)} + H^+_{(aq)}}
\tag{203}
\]
Initial Co + Co’ 0 0.005 - Co’
Final Co + Co’ - x x 0.005 - Co’ + x
\[
K_\mathrm{a}
= \frac{x\,(0.005 - C_0' + x)}{\,C_0 + C_0' - x\,}
\tag{204}
\]
This is solved as usual. The results of the calculations are shown in Table 10.
| Table 2.10 - Effect of adding 0.005 M HCl to buffer solutions from Table 9. Initial concentrations of SO42- and HSO4- are adjusted for the a | |||||||
|---|---|---|---|---|---|---|---|
| Initial [SO42-]1 | Initial [HSO4-] 1 | Initial [SO42-] [HSO4-] | Initial [H+] 1 | x | Equil. [SO42-] | Equil. [HSO4-] | pH |
| 0.0000 | 1.000 | 0 | 4.00E-03 | 9.43E-02 | 0.0943 | 0.9057 | 1.012 |
| 4.90E-03 | 0.9951 | 4.93E-03 | 0.00E+00 | 9.36E-02 | 0.0985 | 0.9015 | 1.033 |
| 0.0859 | 0.9141 | 0.0940 | 0.00E+00 | 5.99E-02 | 0.1458 | 0.8542 | 1.223 |
| 0.4950 | 0.5050 | 0.9802 | 0.00E+00 | 1.00E-02 | 0.5050 | 0.4950 | 2.003 |
| 0.9041 | 0.0959 | 9.427 | 0.00E+00 | 1.07E-03 | 0.9052 | 0.0948 | 2.973 |
| 0.9851 | 0.0149 | 66.11 | 0.00E+00 | 1.53E-04 | 0.9853 | 0.0147 | 3.813 |
| 0.9940 | 6.00E-03 | 165.7 | 0.00E+00 | 6.11E-05 | 0.9941 | 5.94E-03 | 4.213 |
| 1. “New initial” concentrations after adding H+ and reaction with SO42-. | |||||||
| 2. pH calculated after determining changes in concentrations of SO42- and HSO4- by equation [102]. | |||||||
| 3. pH calculated after determining changes in concentrations of SO42- and HSO4- by analogy to equation [96]. (Some of the initial SO42- left over after addition of 0.005 M H+) | |||||||
The results from these last two sets of calculations are compared in Table 11. The pH of a 0.005 M HCl solution alone would be -log 0.005 = 2.3. In this case the buffering capacity becomes poorer as [SO42-]/[HSO4-] exceeds 100. However, the pH is little affected by added strong acid when the ratio decreases below 1. The reasons for the lack of effect of strong acid at low ratios stems from the relatively high value for Ka of HSO4- and the low concentration of added strong acid. At low sulfate/bisulfate ratios bisulfate concentration is almost 1 M, while the added acid is only 0.005 M. Bisulfate is a strong enough (albeit still “weak”) acid that its dissociation is not greatly affected by a small amount of a strong acid, or in other words, sulfate is too weak a base, and the added acid level is too small to significantly affect the sulfate/bisulfate ratio. This sort of effect was not observed for the ammonia/ammonium system where neither Ka nor Kb were exceedingly small.
| Table 2.11 - Comparison of pH of sulfate/bisulfate solutions ([SO42-] + [HSO4-] = 1 M) before and after HCl addition (0.005 M). | ||
|---|---|---|
| Initial [SO42-] / [HSO4-] | pH1 prior to HCl addition | pH1 after HCl addition |
| 0.001 | 1.02 | 1.01 |
| 0.01 | 1.04 | 1.03 |
| 0.1 | 1.23 | 1.22 |
| 1 | 2.01 | 2.00 |
| 10 | 3.00 | 2.97 |
| 100 | 3.99 | 3.81 |
| 1000 | 4.99 | 4.21 |
| 1. pH calculated after determining changes in concentrations of SO42- and HSO4-. | ||
5. The Buffer pH Range of a Weak Acid and its Conjugate Base.
The results from the last two examples show that a solution of a weak acid and its conjugate base manifest significant buffering over a pH range:
\[
0.01 < \frac{[\mathrm{A^-}]}{[\mathrm{HA}]} < 100
\qquad\text{equivalently}\qquad
0.01 < \frac{[\mathrm{B}]}{[\mathrm{BH^+}]} < 100
\tag{205}
\]
The effect is greatest when [A-]/[HA] = 1, and diminishes as the ratio drops or increases. This can be substituted into the expression for pH:
\[
\mathrm{pH}
= \mathrm{p}K_\mathrm{a} + \log(0.01)
\;\;\text{to}\;\;
\mathrm{p}K_\mathrm{a} + \log(100)
\tag{206}
\]
or,
\[
\mathrm{pH} = \mathrm{p}K_\mathrm{a} \pm 2
\tag{207}
\]
This we take to be the normal buffer range for a weak acid and its conjugate base. Within this range pH responds slowly to added strong acid or base, but more so as the ratio [A-]/[HA] diverges from 1.
6. Summary of Buffers.
The preceding material on buffers can be summarized to make the following points for an acid HA and its conjugate base A-:
- This discussion applies only to an acid and its conjugate base!
- Only weak acids/bases exhibit buffering.
- Buffering is most effective when [A-]/[HA] = 1.
- The buffer effect extends mainly over 0.01 < [A-]/[HA] < 100.
- The pH may be estimated from pH = pKa + log [A-]/[HA].
- The equilibrium [A-] and [HA] will be quite close to the initial concentrations as added to the solution, over the range 0.01 < [A-]/[HA] < 100, if one of Ka or Kb is not very small; the assumption becomes less valid as one of Ka or Kb becomes very small (roughly < 10-12).
- The normal buffer range for a weak acid and its conjugate base is pH = pKa ±2
- The equilibrium concentrations can always be calculated from the equilibrium constant expression.
- When Ka > Kb, use the acid dissociation reaction and equilibrium constant to calculate the equilibrium concentrations; when Ka < Kb, use the base dissociation reaction and equilibrium constant.
7. Buffering with Polyprotic Acids/Bases.
Any acid with more than one ionizable proton is polyprotic. In the great majority of cases each successive equilibrium constant is smaller than the preceding one, i.e.
\[
\ce{H_nA <=> H^+ + H_{n-1}A^-}
\tag{208}
\]
\[
\ce{H_{n-1}A^- <=> H^+ + H_{n-2}A^{2-}}
\tag{209}
\]
\[
\ce{H_{n-2}A^{2-} <=> H^+ + H_{n-3}A^{3-}}
\tag{210}
\]
Ka1 > Ka2 > Ka3, etc.
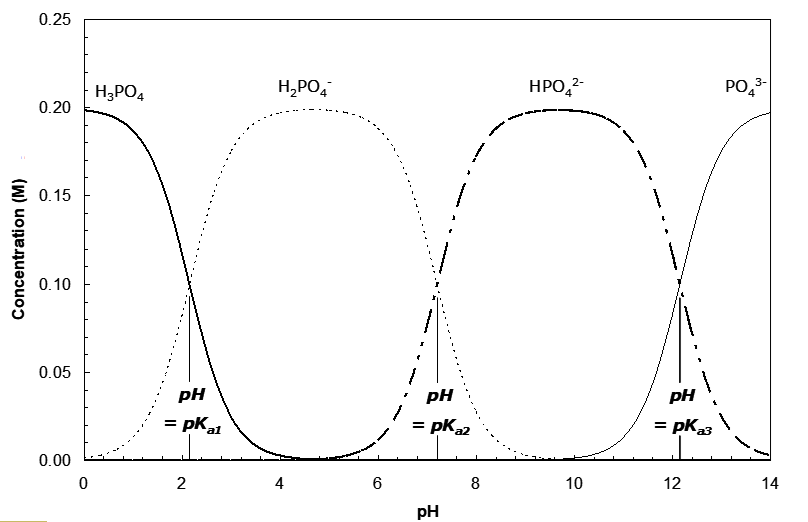
Quite often the Ka values are separated by several orders of magnitude. For an acid H3A, its conjugate base is H2A-; for H2A- it is HA2-, etc. (They differ by one proton, and sometimes an H2O.) We can reasonably expect then that conjugate acid-base pairs derived from polyprotic acids also exhibit buffering. But since there are more numerous species now, might not these make the composition calculations and hence the buffer calculations lot more complicated? The answer is no, so long as the pKa values differ substantially. Consider H3PO4 as an example. The pKa values are:
pKa1 = 2.15; pKa2 = 7.20; pKa3 = 12.15
Ka1 = 1.08 x 10-3; Ka2 = 6.31 x 10-8; Ka3 = 7.08 x 10-13
These differ from each other by a factor of roughly 105. So if we have a solution containing similar concentrations of HPO42- and H2PO4-, both are weak acids and both are weak bases. Hence the reaction,
\[
\ce{H2PO4^-_{(aq)} + H2O_{(l)} <=> H3PO4_{(aq)} + OH^-_{(aq)}}
\tag{211}
\]
\[
K
= \frac{K_\mathrm{b}(\ce{H2PO4^-})}{K_\mathrm{a}(\ce{H3PO4})}
= K_\mathrm{w}
= 1.4 \times 10^{-12}
\tag{212}
\]
occurs to a very small extent. And the reaction,
\[
\ce{HPO4^{2-} <=> H^+ + PO4^{3-}}
\qquad
K_{\mathrm{a3}} = 7.08 \times 10^{-13}
\tag{213}
\]
also occurs to a small extent. Hence very little H3PO4 or PO43- will be present. Thus we can estimate that the pH of a buffer solution having similar concentrations of these two species will be given by,
\[
\mathrm{pH}
= \mathrm{p}K_\mathrm{a}
+ \log\!\left(\frac{[\ce{HPO4^{2-}}]}{[\ce{H2PO4^-}]}\right)
\tag{214}
\]
where the equilibrium concentrations can be taken to be equal to their initial values as added to the solution. Again, the assumption becomes poorer as either Ka or Kb becomes very small, but also if they do not differ greatly. The three buffer points for phosphoric acid then are equal to the pKa values, and for HPO42-/PO43- we can expect that the assumption will be less adequate due to the low Ka for HPO42-. Calculation of compositions for all species in such a system is illustrated in the next section.
4. Speciation Calculations for Polyprotic Acids.
This section demonstrates how to calculate the composition of a complex mixture, specifically a polyprotic acid. The principles can be applied to any problem involving simultaneous equilibria (multiple reactions between related species, all occurring in the same system). The concentrations of chemical species are related to each other by equilibrium constants. All the equilibrium constant expressions must be satisfied simultaneously. When we had just one reaction and equilibrium expression to deal with we could simply set the change in one concentration to be some unknown, account for the change in the others and solve for the unknown using the Ka or Kb expression. That can’t be done in a case where there are multiple simultaneous equilibria. Rather we need to write enough independent equations relating the concentrations of the species so that we can solve them simultaneously. For aqueous H3PO4 we have three such expressions based on the three Ka’s. The concentrations of H+ and OH- are related by Kw. This system, however, contains the following six species: H3PO4, H2PO4-, HPO4 , PO43-, H+ and OH-. We will specify the starting concentration of H3PO4 in solution. If none of the species are assigned particular concentrations, we need six independent equations. We can write the following equation:
\[
[\mathrm{P}]_\text{total}
= [\ce{H3PO4}]
+ [\ce{H2PO4^-}]
+ [\ce{HPO4^{2-}}]
+ [\ce{PO4^{3-}}]
\tag{215}
\]
This is the mass balance for P in all its forms. They all have to add up to what was initially added. If we do not specify one of the other concentrations then we need one more. Often charge balance can be used. The idea is that the sum of the concentrations of the positive charges must equal the sum of the concentrations of the negative charges. For instance if we have y M of PO43- in solution, then the concentration of negative charge due to PO43- is 3y.
\[
[\ce{H^+}]
= [\ce{OH^-}]
+ [\ce{H2PO4^-}]
+ 2[\ce{HPO4^{2-}}]
+ 3[\ce{PO4^{3-}}]
\tag{216}
\]
Thus we would have the three Ka expressions, Kw, the mass balance on P and the charge balance equation. With these six we could determine the concentrations of all species for a given concentration of H3PO4.
For the purposes of this example, however, we will specify the solution pH in order to see how composition varies with pH. This will further elucidate the discussion about buffering for polyprotic acids. Having this makes the charge balance unnecessary. Having pH also necessarily determines [OH-] through Kw. This leaves:
\[
\ce{H3A <=> H^+ + H2A^-}
\qquad
K_{\mathrm{a1}}
= \frac{[\ce{H^+}][\ce{H2A^-}]}{[\ce{H3A}]}
\tag{217}
\]
\[
\ce{H2A^- <=> H^+ + HA^{2-}}
\qquad
K_{\mathrm{a2}}
= \frac{[\ce{H^+}][\ce{HA^{2-}}]}{[\ce{H2A^-}]}
\tag{218}
\]
\[
\ce{HA^{2-} <=> H^+ + A^{3-}}
\qquad
K_{\mathrm{a3}}
= \frac{[\ce{H^+}][\ce{A^{3-}}]}{[\ce{HA^{2-}}]}
\tag{219}
\]
We need to eliminate variables. Express of all ‘A’ concentrations (A = PO4) in terms of [H3A]:
\[
[\ce{H2A^-}]
= \frac{K_{\mathrm{a1}}[\ce{H3A}]}{[\ce{H^+}]}
\tag{220}
\]
\[
[\ce{HA^{2-}}]
= \frac{K_{\mathrm{a2}}[\ce{H2A^-}]}{[\ce{H^+}]}
= \frac{K_{\mathrm{a1}}K_{\mathrm{a2}}[\ce{H3A}]}{[\ce{H^+}]^{2}}
\tag{221}
\]
\[
[\ce{A^{3-}}]
= \frac{K_{\mathrm{a3}}[\ce{HA^{2-}}]}{[\ce{H^+}]}
= \frac{K_{\mathrm{a1}}K_{\mathrm{a2}}K_{\mathrm{a3}}[\ce{H3A}]}{[\ce{H^+}]^{3}}
\tag{222}
\]
Substituting into the mass balance equation:
\[
[\mathrm{P}]_\mathrm{T}
= [\ce{H3A}]
+ \frac{K_{\mathrm{a1}}[\ce{H3A}]}{[\ce{H^+}]}
+ \frac{K_{\mathrm{a1}}K_{\mathrm{a2}}[\ce{H3A}]}{[\ce{H^+}]^{2}}
+ \frac{K_{\mathrm{a1}}K_{\mathrm{a2}}K_{\mathrm{a3}}[\ce{H3A}]}{[\ce{H^+}]^{3}}
\tag{223}
\]
If we specify a total phosphoric acid concentration and the pH, this equation is readily solved for [H3A]. A plot of the concentrations of the various species as a function of pH is shown below. What is apparent is that where the curves for two species pass (e.g. H3PO4 and H2PO4-; H2PO4- and HPO42-; HPO42- and PO43-) there is very little of any other species present. The three points where the lines cross correspond to the buffer points for the three acids (the pKa’s). In general, for a weak polyprotic acid, the pKa values are the buffer points for the acids and their conjugate bases. And again, the buffer regions range over 0.01 < pKai < 100. In cases where the pKa’s are not so widely separated, there may be significant concentrations of species other than those that define the buffer points.
5. Buffers Based on “Salts” of Weak Acids and Weak Bases.
When a strong acid is reacted with a strong base, a salt is formed, e.g.
\[
\ce{HCl_{(aq)} + NaOH_{(aq)} -> NaCl_{(aq)} + H2O_{(l)}}
\tag{224}
\]
The salt, NaCl, is composed of an exceedingly weak acid and an exceedingly weak base. When dissolved in pure water, this salt solution has a pH of very nearly 7; neutral pH.
But when a weak acid and a weak base react a compound is produced that may or may not be a salt per se. For example ammonium sulfide, nominally (NH4)2S, is obtained when 2 molar equivalents of H2S gas is bubbled into 1 molar equivalent of aqueous NH3. But Kb (S2-) ≈ 3.2 x 104 (quite a strong base) whereas Kb (NH3) is much smaller (1.74 x 10-5), so discrete S2- ions are not present in “ammonium sulfide.” The “salt” is actually a combination of NH3 and NH4+HS-.
An important example of a weak acid-weak base salt in hydrometallurgy is ammonium carbonate:
\[
\ce{2NH3_{(aq)} + H2O_{(l)} + CO2_{(aq)} -> (NH4)2CO3_{(aq)}}
\tag{225}
\]
The compound can be precipitated as a solid from solution, although it readily loses ammonia. This compound is important in some kinds of nickel leaching processes (the Caron process), allowing [Ni(NH3)6]+2 to form, and its solutions are buffers. Although it is not a conjugate acid-base pair, it is still a weak base plus a weak acid. The buffer effect can be quantitatively demonstrated by applying the speciation methods developed above. We might first think that since NH4+ is an acid and CO32- is a base, the relevant equilibrium expressions would be for acid dissociation of NH4+ and base dissociation of CO32-. However, this makes the calculations more complicated, and we would ultimately have to express either [OH-] in terms of [H+] or vice versa anyway. It is simpler to start with equilibria involving either H+ only or OH- only. Finally, both CO2 and NH3 can volatilize from solution. In an open vessel this would continue because no equilibrium CO2 or NH3 pressure would be established above the solution; they would both waft away. In a closed system equilibrium pressures of these two gases would develop and this would properly have to be taken into account. For the sake of this discussion we will ignore this, though it is not hard to incorporate.
\[
\ce{NH4^+ <=> H^+ + NH3_{(aq)}}
\qquad
K_{\mathrm{aN}}
= [\ce{H^+}][\ce{NH3}]
\tag{226}
\]
\[
\ce{NH4^+ + CO2_{(aq)} + H2O <=> H^+ + HCO3^-}
\qquad
K_{\mathrm{a1}}
= [\ce{H^+}][\ce{HCO3^-}]
\tag{227}
\]
\[
\ce{HCO3^- <=> H^+ + CO3^{2-}}
\qquad
K_{\mathrm{a2}}
= [\ce{H^+}][\ce{CO3^{2-}}]
\tag{228}
\]
\[
K_\mathrm{w}
= [\ce{H^+}][\ce{OH^-}]
\tag{229}
\]
The species to account for are: NH4+, NH3 aq, CO32-, HCO3-, CO2 (aq), OH- and H+. We have seven species and four equations so far. We can write mass balance equations for total ammonia and for total CO2. Let the initial concentration of ammonium carbonate, (NH4)2CO3, be Co M. At equilibrium,
\[
[\ce{NH3}] + [\ce{NH4^+}] = 2C_0
\tag{230}
\]
\[
[\ce{CO2}] + [\ce{HCO3^-}] + [\ce{CO3^{2-}}] = C_0
\tag{231}
\]
We can also write a charge balance equation:
\[
[\ce{H^+}] + [\ce{NH4^+}]
= [\ce{OH^-}]
+ [\ce{HCO3^-}]
+ 2[\ce{CO3^{2-}}]
\tag{232}
\]
The objective will be to demonstrate that this solution is a buffer. If so, the addition of a small amount of a strong acid or base will not significantly change the pH. Let the concentration of added NaOH = y. (When y = 0 we have simply the ammonium carbonate solution alone.) The acids in solution are NH4+, CO2 and HCO3-. For the purposes of calculating the equilibrium composition we can treat this as a solution containing new initial concentrations of NH3 and NH4+ due the reaction,
\[
\ce{NH4^+ + OH^- -> NH3 + H2O}
\qquad
K = \frac{1}{K_\mathrm{b}} \gg 1
\tag{233}
\]
Or we could assume that one of CO2 or HCO3- is set to new initial concentrations by reaction with OH-. Adjusted initial concentrations of NH4+ and NH3 are convenient, though the others could be used. This is equivalent to a solution containing 2Co - y M NH4+, y M NH3, y M Na+ and Co M of CO32-. None of the equilibrium expressions are changed. Neither are the mass balance expressions; they are still true as written. The only thing that changes is the charge balance equation. That now must incorporate [Na+] = y:
\[
[\ce{H^+}] + [\ce{NH4^+}] + y
= [\ce{OH^-}]
+ [\ce{HCO3^-}]
+ 2[\ce{CO3^{2-}}]
\tag{234}
\]
We have to specify values for Co and y. Now the composition can be determined and we can see what effect added NaOH has on the pH.
Variables need to be eliminated. The [NH3] can be expressed in terms of [NH4+] using KaN. This can be put into the ammonia mass balance equation:
\[
[\ce{NH3}] + [\ce{NH4^+}]
= 2C_0
= [\ce{NH4^+}]
+ \frac{K_{\mathrm{aN}}[\ce{NH4^+}]}{[\ce{H^+}]}
\tag{235}
\]
\[
[\ce{NH3}] + [\ce{NH4^+}]
= 2C_0
= [\ce{NH4^+}]
+ \frac{K_{\mathrm{aN}}[\ce{NH4^+}]}{[\ce{H^+}]}
\tag{236}
\]
The concentrations of the three carbon species can be expressed in terms of one of them. It does not really matter which, though CO32- or CO2 are more convenient.
\[
[\ce{HCO3^-}]
= \frac{[\ce{H^+}][\ce{CO3^{2-}}]}{K_{\mathrm{a2}}}
\tag{237}
\]
\[
[\ce{CO2}]
= \frac{[\ce{H^+}][\ce{HCO3^-}]}{K_{\mathrm{a1}}}
= \frac{[\ce{H^+}]^{2}[\ce{CO3^{2-}}]}{K_{\mathrm{a1}}K_{\mathrm{a2}}}
\tag{238}
\]
\[
[\ce{OH^-}]
= \frac{K_\mathrm{w}}{[\ce{H^+}]}
\tag{239}
\]
From the charge balance equation:
\[
[\ce{NH4^+}]
= [\ce{OH^-}]
+ [\ce{HCO3^-}]
+ 2[\ce{CO3^{2-}}]
- [\ce{H^+}]
- y
\tag{240}
\]
Expressions for the concentrations may be substituted into this equation:
\[
\frac{2C_0}{1 + \dfrac{K_{\mathrm{aN}}}{[\ce{H^+}]}}
=
\left(
2[\ce{CO3^{2-}}]
+ \frac{[\ce{H^+}][\ce{CO3^{2-}}]}{K_{\mathrm{a2}}}
+ \frac{K_\mathrm{w}}{[\ce{H^+}]}
- [\ce{H^+}]
- y
\right)
\tag{241}\]
This equation has only two variables: [CO32-] and [H+]. We can relate [CO32-] to [H+] through the mass balance on CO2,
\[
[\ce{CO2}] + [\ce{HCO3^-}] + [\ce{CO3^{2-}}] = C_0
\tag{242}
\]
and substituting in expressions for [CO2] and [HCO3-].
\[
[\ce{CO3^{2-}}]
+ \frac{[\ce{CO3^{2-}}][\ce{H^+}]}{K_{\mathrm{a2}}}
+ \frac{[\ce{CO3^{2-}}][\ce{H^+}]^{2}}{K_{\mathrm{a1}}K_{\mathrm{a2}}}
= C_0
\tag{243}
\]
Rearranging gives:
\[
[\ce{CO3^{2-}}]
= \frac{C_0}{
1
+ \dfrac{[\ce{H^+}]}{K_{\mathrm{a2}}}
+ \dfrac{[\ce{H^+}]^{2}}{K_{\mathrm{a1}}K_{\mathrm{a2}}}
}
\tag{244}
\]
Substituting this for [CO32-] in the charge balance equation and rearranging finally gives:
\[
\frac{
\left(
2 + \frac{[\ce{H^+}]}{K_{\mathrm{a2}}}
\right)
\left(
\frac{C_0}{
1
+ \dfrac{[\ce{H^+}]}{K_{\mathrm{a2}}}
+ \dfrac{[\ce{H^+}]^{2}}{K_{\mathrm{a1}}K_{\mathrm{a2}}}
}
\right)
\left(
1 + \frac{K_{\mathrm{aN}}}{[\ce{H^+}]}
\right)
+
\left(
\frac{K_\mathrm{w}}{[\ce{H^+}]} - [\ce{H^+}]
\right)
\left(
1 + \frac{K_{\mathrm{aN}}}{[\ce{H^+}]}
\right)
}{2C_0}
= 1
\tag{245}
\]
This can now be solved for [H+], and after that [CO32-] can be found, then the rest of the values. The best way to solve the above equation is to use iteration. A provisional value for [H+] is set. Carbonate is a stronger base than NH4+ is an acid, based on Kb and Ka, respectively, so value >10-7 is required; 10-9 is a reasonable guess. Using this value, the number calculated from equation (245) ≠ 1. (For Co = 0.5 M and no added NaOH, setting [H+] = 10-9 yields a value of 0.82 from equation (245)). Multiply the provisional [H+] by the result and recalculate (e.g. 0.82 x 10-9, using the preceding example). Multiply the second [H+] by the new result from the above equation. Repeat the process until the calculated result is very close to 1. The calculations work for solutions with and without added NaOH.
The original question was whether such a solution is a buffer. The pH values of a 0.5 M ammonium carbonate solution with and without added NaOH are shown in the table below. Clearly the solution strongly attenuates the change in pH that would occur due to addition of strong base. The same effect would be manifest for addition of strong acid.
| Table 2.12 - pH of a 0.5 M (NH4)2CO3 solution and of the same solution with added NaOH. The pH of solutions containing NaOH alone are provided for comparison. Added NaOH M pH pH of NaOH solution alone | ||
|---|---|---|
| Added NaOH M | pH | pH of NaOH solution alone |
| 0 | 9.18 | - |
| 0.05 | 9.26 | 12.7 |
| 0.1 | 9.34 | 13.0 |
| 0.2 | 9.49 | 13.3 |
Media Attributions
- Ch0-2_F1_pKw_Water_Dissociation_Temps © Bé Wassink and Amir M. Dehkoda is licensed under a CC BY-NC (Attribution NonCommercial) license
- Ch0-2_F2_HCl_Activity_v_Molality © Bé Wassink and Amir M. Dehkoda is licensed under a CC BY-NC (Attribution NonCommercial) license
- Ch0-2_F3_CN_Fraction_v_pH © Bé Wassink and Amir M. Dehkoda is licensed under a CC BY-NC (Attribution NonCommercial) license
- Ch0-2_F4_Speciation_Diagram_H3PO4 © Bé Wassink and Amir M. Dehkoda is licensed under a CC BY-NC (Attribution NonCommercial) license
A unit of concentration. 1 molal = 1 mole of specified solute per kg of solvent. Symbol: m. This is the common unit for concentration in thermodynamics. (Molarity is moles of solute per litre of solution. This is operationally convenient and thermodynamically inconvenient, since volume varies with temperature. Hence so does molarity. Molality does not vary with temperature.)
A concentration unit, usually mg (milligrams) of species of interest per kg of total mass of the sample. Since the density of water is nearly 1000 g/L (998 g/L at 21°C), and the density of a dilute solution is close to that of water, ppm is also often used synonymously with mg/L, though this is not strictly correct.
