Chapter IV: Introduction to Leaching
2. Leaching and Process Chemistry
This is the overarching chemical consideration for all hydrometallurgy processes. And it is usually leaching that determines the chemical reaction medium for the overall process. First consider the thermodynamic possibilities. This is often done with the aid of Eh-pH diagrams and associated basic chemistry.
General Classes of Leaching Reactions
There are four possible things we can do to try to leach something in hydrometallurgy:
- Dissolve in water
- Dissolve in acid or base (adjust pH)
- Oxidize or reduce it
- Form complexes with suitable ligands (complexants)
Combinations of these may be necessary. The more that has to be done, the greater the complexity, and the higher the cost. The possibilities are outlined in the figure below.
- Soluble in water. Leaching in hydrometallurgy involves water by definition. If a mineral is readily soluble in water, e.g. CuSO4·5H2O (calcanthite), then that is all that is needed. This is rare.
- Soluble in acid. Some minerals require only acid to dissolve.
- Soluble in base. Some minerals dissolve in basic solution, e.g. NaOH. Some may be soluble in either acid or base; they are amphoteric. In general base is more expensive than acid.
- Can be oxidized. Where a mineral cannot be readily dissolved in acid or base alone, oxidation may be possible.
- Can be reduced. Few minerals can be treated in this way. Reducing agents are more costly than oxidants, in general.
- The mineral can be dissolved by forming a complex, usually in acidic or basic solution.
- A combination a complexant and an oxidant in acid or base, or much less likely, complexant and reductant in acid or base may be needed.
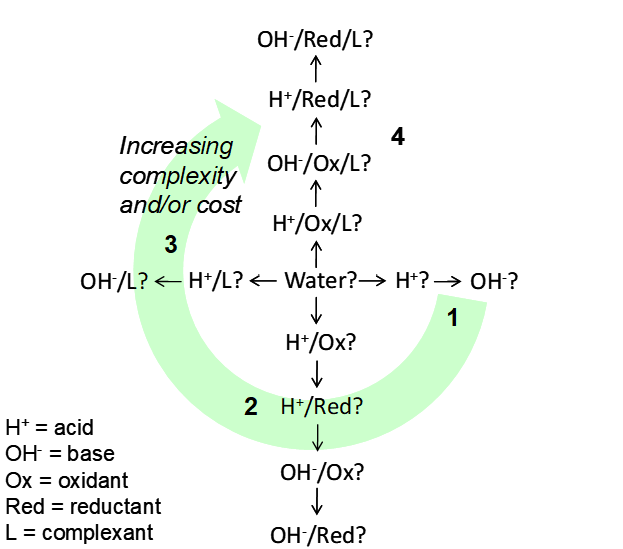
Illustrations of How to Choose a Leaching Medium
First consider the nature of the materials to be leached. Based on the solubility rules for salts, most oxides, hydroxides and sulfides are insoluble in water. This is important, because many of the minerals of interest are oxides, hydroxides or sulfides. For any other mineral classes the same types of considerations apply. Native metals (e.g. Au, Cu) are insoluble in water; they dissolve only by corrosion. Some examples of how suitable leaching chemistry can be worked out from basic principles are provided below.
Leaching of CuO and CuS
Based on the Ksp values for mineral compounds, many oxides and hydroxides are soluble in acid at low, but practically attainable pH. Consider, for example, dissolution of CuO in acid:
\[\ce{CuO(s) + H_2O(l) -> Cu^{2+}(aq) + 2OH^-(aq)} \quad K_{sp} = 2.2 \times 10^{-21} \tag{1}\]
\[\ce{2OH^-(aq) + 2H^+(aq) -> 2H_2O(l)} \quad K = (1/K_w)^2 = 1 \times 10^{28} \tag{2}\]
\[\ce{CuO(s) + 2H^+(aq) -> Cu^{2+}(aq) + H_2O(l)} \quad K_l = K_{sp}/K_w^2 = 2.2 \times 10^{7} \tag{3}\]
The overall reaction is very favourable and CuO can be readily dissolved in acid solution. This is confirmed by considering the CuO/Cu+2 vertical line in the Eh-pH diagram for the Cu-H2O system shown in Figure 2.2.
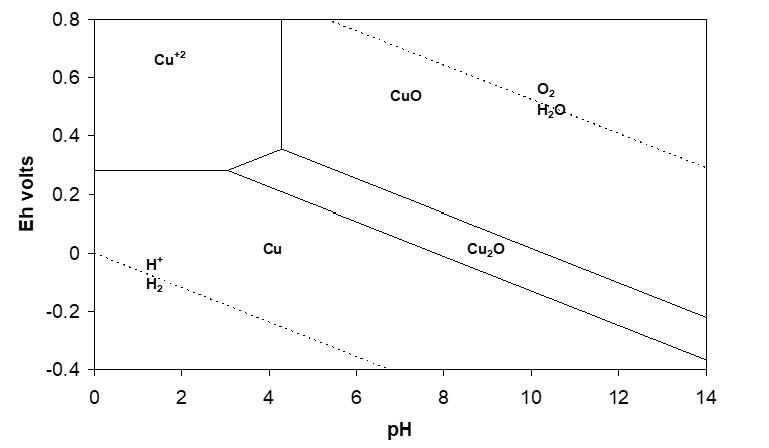
The next question is how soluble is CuO at a given pH? This can be readily determined from the equilibrium constant expression:
\[\ce{K = \frac{[Cu^{2+}]}{[H^{+}]_2}} \]
\[\ce{log[Cu^{2+}] = log K_1 - 2 pH} \]
(The expression is readily derived from the expression for pH of a vertical line used to draw Eh-pH diagrams.) In this example, at pH 3.67 the solubility of Cu+2 will be around 1 M (actually, the activity will be 1 M, and since such solutions are highly non-ideal, the solubility will be less than this). Obviously the lower Kl, the less soluble the mineral is at a given pH. Note that the slope of the log solubility vs. pH plot depends on the moles H+/mol metal ion dissolved according to the chemical reaction. What drives such reactions is the strong favourability for formation of water.
We did not pay attention to Cu2O, which appears in the Eh-pH diagram. Since it lies below CuO, it would form only if we were to reduce a Cu(II) species (either [latex]Cu^{+2} \displaystyle[/latex] or CuO). If we leach in the open air, the presence of oxygen ensures that we maintain a high enough potential (NB E°O2/H2O = 1.23 V) that reducing conditions do not arise. (On the other hand, if for some reason we were to see Cu2O forming, it would be telling us that some adventitious species present in the system was generating a reducing environment.)
In general, sulfides are less soluble in water than oxides/hydroxides, and, H2S is a much less favourable product after reaction with acid. Mostly, in such cases we need to be able to oxidize the sulfide portion of the mineral, either to elemental sulfur, or some other higher oxidation state form (usually sulfate). Consider the Eh-pH diagram for the Cu−S−H2O system in Figure 2.3. From this diagram, it is clear that the only possibility for leaching CuS is in acid solution and with the use of an oxidant.
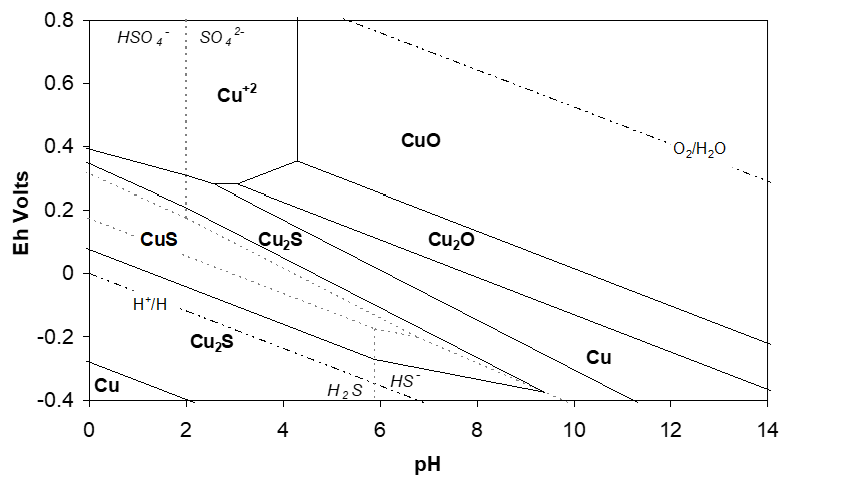
Oxygen is suitable oxidant, based on the fact that the O2/H2O reduction potential (Eh) is greater than that required to sustain Cu+2 in solution over the whole of the region where Cu+2 is stable. And these considerations are verified by the following reactions:
\[\ce{CuS(s) -> Cu^{2+}(aq) + S^{2-}(aq)} \quad K_{sp} = 8 \times 10^{-37} \tag{4}\]
\[\ce{CuS(s) + 2H^+(aq) -> Cu^{2+}(aq) + H_2S(g)} \quad K_l = 1.1 \times 10^{-15} \tag{5}\]
\[\ce{CuS(s) + 2O_2(g) -> Cu^{2+}(aq) + SO_4^{2-}(aq)} \quad K'_l = 4 \times 10^{109}\;(!) \tag{6}\]
To this point thermodynamics is serving us accurately. Only oxidation in acid solution can be used to leach CuS. But now we come up against the discrepancy between thermodynamics and kinetics that is rather common in hydrometallurgy; for instance, oxygen alone reacts very slowly, particularly near room temperature with many minerals, including with CuS. However, bacteria of the family thiobacillus occur naturally and are part of the earth's natural sulfur cycle. They can be readily adapted for use in leaching, and they quite efficiently promote oxidation of several copper sulfide minerals (and others). They oxidize the sulfide right through to sulfate, forming sulfuric acid in the process. Thus bioleaching, as it is called, is commonly practiced for some copper sulfide minerals in low grade ores (especially those from which a concentrate cannot be readily made; if a concentrate can be made pyrometallurgical methods are more likely to be employed).
Continuing with this theme, ferric ion is often used as a "surrogate" oxidant to help overcome the inherent slowness of O2 as an oxidant:
\[\ce{Au^+(aq) + e^- -> Au(s)} \quad E^\circ = 1.69\;\text{V} \tag{7}\]
Ferric ions are often available in the ore from oxidation of pyrite, i.e. FeS2, or other iron minerals. Ferric ion can oxidize many sulfide minerals (e.g. CuS) to form elemental sulfur and aqueous Mn+ cation (e.g. Cu+2). In addition, a strain of bacteria named thiobacillus ferrooxidans readily catalyzes oxygen oxidation of ferrous ion to ferric. This is used to regenerate ferric ion. Then the ferric/ferrous couple is used as a redox catalyst in the oxidative leaching process. To summarize:
\[\ce{CuS(s) + 2Fe^{3+}(aq) -> Cu^{2+}(aq) + 2Fe^{2+}(aq) + S(s)} \tag{8}\]
\[\ce{2Fe^{2+}(aq) + 1/2O_2(g) + 2H^+(aq) -> 2Fe^{3+}(aq) + H_2O(l)} \tag{9}\]
(By thiobacillus ferrooxidans)
\[\ce{S(s) + 3/2O_2(g) + H_2O(l) -> H_2SO_4(aq)} \tag{10}\]
(By thiobacillus thiooxidans and ferrooxidans)
Ferric ion is also used as a surrogate oxidant to catalyze O2 oxidation of metal sulfides at higher temperature and pressures, such as in an autoclave. A good example is sphalerite concentrate leaching (ZnS), which is practiced using autoclaves at −150°C and under oxygen pressure. Ferric ion catalyzes the O2 oxidation process, which would otherwise be too slow, even under autoclave conditions. However, now due to the higher temperature, ferrous can be readily reoxidized to ferric by oxygen. (Bacteria do not survive under autoclave conditions.) The same applies to many copper sulfides, but to date the economics of autoclave leaching have not been favourable.
Considering the Eh−pH diagram for the Cu-S-H2O system alone might be confusing. As far as thermodynamics is concerned, ferric ion is sufficiently strongly oxidizing to convert CuS into Cu+2 and HSO4-, say at pH −1. Yet when we react Fe+3 with CuS at −25°C, we find that CuS does leach, but that Cu+2 and solid sulfur, S, are formed, and not HSO4−(in the absence of sulfur-oxidizing bacteria). Nothing on the diagram suggests that this would occur. Nor do we see that Cu2S forms. In fact, if we start with Cu2S the first thing that happens is,
\[\ce{Cu_2S(s) + 2Fe^{3+}(aq) -> Cu^{2+}(aq) + 2Fe^{2+}(aq) + CuS(s)} \tag{11}\]
It is imperative that we understand what the Eh-pH diagram is, and is not telling us. In general thermodynamics tells us what is possible; that a reaction may or may not be spontaneous (occur naturally, on its own, without energy input), or that if it is not spontaneous, what we have to do to make it go (input some form of energy). What it does not tell us is the intermediate steps or reactions that may be involved in getting from some initial state (e.g. CuS + ferric sulfate at pH 1 and 25°C) to some final state (such as Cu+2aq, HSO4−aq, Fe+2aq, pH 1, 25°C). That is the domain of kinetics. Thermodynamics does not care if it takes a millisecond or a million years. In the limit of long enough time, it simply tells us what will occur. An Eh−pH diagram is even more constrained. It tells us what the most stable state is for some combination of elements (for our purposes in water) under specified conditions of pH and electrochemical potential*, e.g. Cu, S and H2O.
And how do we maintain some specified set of conditions? Acids or bases are used to adjust pH; a redox couple can be used to maintain an electrochemical potential (likewise an electrical power supply attached to an electrochemical cell). The pH control seems easy enough, but the latter works in much the same way. Recall the Nernst equation, as illustrated below for the Fe+3/Fe+2 couple:
\[\ce{E = E^{\circ} - \frac{2.303RT}{F} \log \frac{a_{Fe^{2+}}}{a_{Fe^{3+}}}}\]
An electrochemical potential can be maintained somewhere near E° (about 0.68 V in sulfuric acid solution) by maintaining a fixed ratio of ferrous:ferric ion activities. Now, note the similarity to the equation for the pH of a weak acid:
\[\ce{pH = pK_a + log \frac{a_{A^-}}{a_{HA}}}\]
where A- and HA are a conjugate acid base pair. Substantial and similar concentrations of an oxidant and its corresponding reductant (a redox couple) may act as an electrochemical potential buffer, just as an acid and its conjugate base can act as a pH buffer (controlling the pH near pKa). The additional complexity in the case of electrochemical potential is that for a given reaction and set of conditions, many redox couples react very slowly, while others react rapidly. Those that react rapidly do establish, or bring into effect the electrochemical potential that thermodynamics says they will. Those that react slowly do not do so in a practical period of time; kinetics inhibits. Knowing which situation pertains is a matter of experience, though there are good chemical reasons. It is more common (though not universally so) that acids and bases react quite rapidly to establish the expected pH.
not tell us is that there are numerous other possible half reactions and thus chemical reactions. Some of the possibilities are qualitatively summarized in the figure below. The two reduction half reaction couples indicated in bold correspond to lines on the Eh−pH diagram in Figure 2.3. The other Cu/S reduction half reaction couples do not appear on the diagram; neither does the Fe+3/Fe+2 couple. The Eh for each couple would be the value for some specified pH < 2, where HSO4− is the dominant S(VI) species. Double-headed arrows at right indicate the ΔE (cell potential) for ferric oxidation of Cu/S species. (The Fe+2/Fe+3 activity ratio would have some specified value, the exact value of which is not important here. What matters here is that EhFe+3/Fe+2 lies above the other couples in the diagram.
What it does not tell us is that there are numerous other possible half reactions and thus chemical reactions. Some of the possibilities are qualitatively summarized in the figure below. The two reduction half reaction couples indicated in bold correspond to lines on the Eh-pH diagram in Figure 3. The other Cu/S reduction half reaction couples do not appear on the diagram; neither does the Fe+3/Fe+2 couple. The Eh for each couple would be the value for some specified pH < 2, where HSO4- is the dominant S(VI) species. Double-headed arrows at right indicate the DE (cell potential) for ferric oxidation of Cu/S species. (The Fe+2/Fe+3 activity ratio would have some specified value, the exact value of which is not important here. What matters here is that EhFe+3/Fe+2 lies above the other couples in the diagram.)
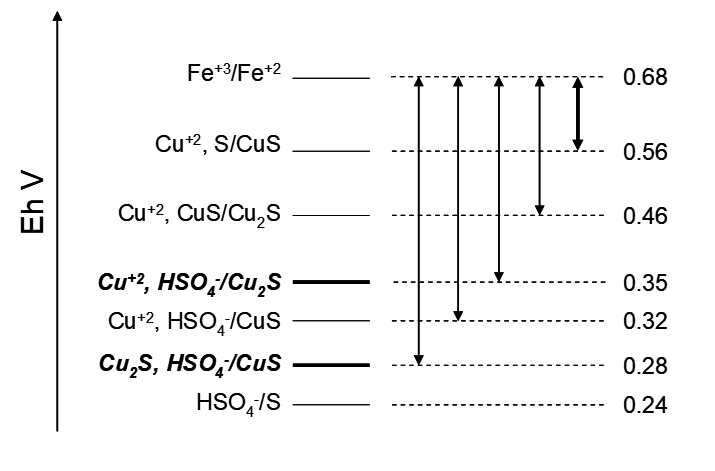
From this diagram and the Eh-pH diagram we can see that Fe+3 is a strong enough oxidant to be able to spontaneously oxidize CuS into Cu2S and Cu2S into Cu+2 and HSO4−. These two half reactions represent the boundaries between the CuS/Cu2S and Cu2S/Cu+2, HSO4−regions, respectively. And since these are the thermodynamically most stable species for the relevant Eh-pH domains, they are the only ones depicted on the Eh-pH diagram. But, as we have already noted, ferric oxidations of CuS to Cu2S + HSO4- and of Cu2S to Cu+2 + HSO4- are exceedingly slow. Likewise, the thermodynamically favourable oxidation of CuS to Cu+2 and HSO4- (as suggested by the Eh-pH diagram) by Fe+3 occurs only exceedingly slowly. Although other half reactions depicted in Figure 2.4 do not appear as boundaries between regions on the Eh-pH diagram, they are otherwise perfectly valid half reactions. Thus Cu2S can be oxidized by Fe+3 to CuS and Cu+2, and CuS can be oxidized to Cu+2 and S; the cell voltages (e.g. ΔE = EFe+3/Fe+2 –EhCu+2, CuS/Cu2S = 0.68 – 0.46 = 0.2 V) are positive, and therefore the reactions are favourable. Finally, note that while the oxidation of S to HSO4- by Fe+3 is quite favourable, this reaction is exceedingly slow. Thus an Eh-pH diagram, while a good place to start, does not provide any information about kinetics, and kinetics may obviate some favourable transformations, even perhaps the most favourable ones. Then other reactions may actually occur in practice, and these will still be thermodynamically favourable, even if not the most favourable.
Finally, note too then that an Eh-pH diagram provides no certain information on the steps through which a reaction can occur. Looking at the Cu-S-H2O diagram we might naively expect that ferric leaching of CuS would first from Cu2S, then Cu+2 + HSO4-. In fact this does not occur.
Media Attributions
- Ch4_F1_Leaching_Chemical_Conditions © Bé Wassink and Amir M. Dehkoda is licensed under a CC BY-NC (Attribution NonCommercial) license
- Ch4_F2_0.01m_25C_CuH2O_Eh-pH © Bé Wassink and Amir M. Dehkoda is licensed under a CC BY-NC (Attribution NonCommercial) license
- Ch4_F3_0.01m_25C_CuSH2O_Eh-pH © Bé Wassink and Amir M. Dehkoda is licensed under a CC BY-NC (Attribution NonCommercial) license
- Ch4_F4_Cu_S_Half_Reaction_Potentials © Bé Wassink and Amir M. Dehkoda is licensed under a CC BY-NC (Attribution NonCommercial) license
An autoclave is a machine that uses steam under pressure to kill harmful bacteria, viruses, fungi, and spores on items that are placed inside a pressure vessel.
