Chapter III: Introduction to Eh pH Diagrams
2. Review of Basic Electrochemistry
Before we can delve into Eh-pH diagrams, the associated electrochemistry has to be in place. If this material is familiar, it may be skipped.
2.1 Electron Transfer Reactions
Many (though not all) reactions involve transfer of electrons between reagents. These may be called electron transfer reactions, redox reactions or electrochemical reactions (the latter refers specifically to processes occurring at electrodes). In normal chemical systems the electron does not exist outside of being attached to atoms, i.e. free electrons do not survive. The energy of a free electron would be so high that it would react with the first thing it encounters. The transfer of electrons from one chemical species to another is a means of transferring energy. Electrons moving from one species to another are essentially energy in transit, just as heat is energy in transit.
There are two reasons that electrons transfer between chemical species. One is that the system (the chemical reaction) and its surroundings together have higher net entropy than the reactants - a spontaneous chemical reaction.* The electrons in one reactant (the reducing agent) are sufficiently energetic to be spontaneously accepted by another reactant (the oxidizing agent). The second reason is that we force an unfavourable electron transfer reaction by imparting extra energy to the electrons, by means of an external applied voltage. This is the basis for electrolysis.
Further, it can be seen that we can consider an electron transfer reaction to be comprised of two parts - something getting reduced, and something getting oxidized. This is the only way that electron transfer reactions can occur. The electrons must be taken from one reagent and accepted by another. (Again, free electrons do not exist.) Thus we can separate an electron transfer reaction into two half reactions, one a reduction and one an oxidation. It must be borne in mind that half reactions never occur in isolation; the electrons must come from one reactant be delivered to another.
2.2 The Electrochemical Potential
This is actually a quite straightforward quantity. It is simply the tendency of a chemical species to accept electrons and be reduced under the specified conditions (like pressure, temperature, activities of reactants and products). The higher the potential, the stronger the driving force for this to occur. Now we are specifically focusing on a reduction half reaction, e.g.
\[\ce{Cu^{2+}_{aq} + 2e^- = Cu_s} \tag{1}\]
Recall that what drives chemical reactions (makes them spontaneous) is that over the system and its surroundings there is a net increase in entropy. Metaphorically, entropy can be thought of as the concentration of energy. It is intuitively reasonable that concentrated energy tends to naturally disperse; degrades to a lower "concentration." The Gibbs free energy equation reflects this link between entropy and spontaneity. Since
ΔG = ΔH - T ΔS at some specified temperature T,
\[\ce{\frac{-\Delta G}{T} = \frac{-\Delta H}{T} + \Delta S}\]
ΔH is the enthalpy change for the system. Then -ΔH is the heat flow to the surroundings. At constant pressure ΔH is the heat flow into the system. Then
-ΔH/T = -qΡ/T (qΡ being heat flow at constant pressure) and this has the form of the entropy. It can be shown (see suitable thermodynamics textbooks) that in fact
-ΔH/T is the entropy change of the surroundings. Thus,
\[\ce{-\Delta G/T = \Delta S_{surroundings} + \Delta S_{system} = Total\ \Delta S}\]
This is the basis upon which we say that a reaction is spontaneous; if ΔG < 0. It applies to all chemical processes.
The Cu+2 ion in aqueous solution has some potential to accept an electron and be reduced to Cu. If we can measure that electrochemical potential for any conceivable half reaction, we can understand and combine those half reactions to perform chemical transformations. The problem though is that there is no way to measure the reduction potential for a half reaction in isolation; electron transfer reactions require a reactant source of electrons and a reactant to accept them. For instance,
\[
\begin{align*}
&\ce{Cu^{2+}_{aq}}+\ce{2e-}=\ce{Cu_s}\tag{2} \\
&\ce{H2_g}=\ce{2H+_{aq}}+\ce{2e-}\tag{3} \\
\hline
\text{net: }&\ce{Cu^{2+}_{aq}}+\ce{H2_g}=\ce{Cu_s}+\ce{2H+_{aq}}\tag{4}
\end{align*}
\]
Hydrogen gas is the source of the electrons; Cu+2 accepts them. (It is the same in electrolysis reactions, which are thermodynamically unfavourable and are forced to go by imposition of a voltage; the electrons are forcibly taken from one reactant and forced onto another.) But, now we have two half reactions that are combined into an overall reaction. We can measure the potential difference (voltage) that is manifested between the two half cells as illustrated in the figure below. This is the measure of the driving force for the electrons to transfer as per the reaction. But, electron transfer occurs. It does not undergo chemical transformation. The salt bridge allows the electrical circuit to be completed by ionic conduction between compartments, otherwise no current would flow.
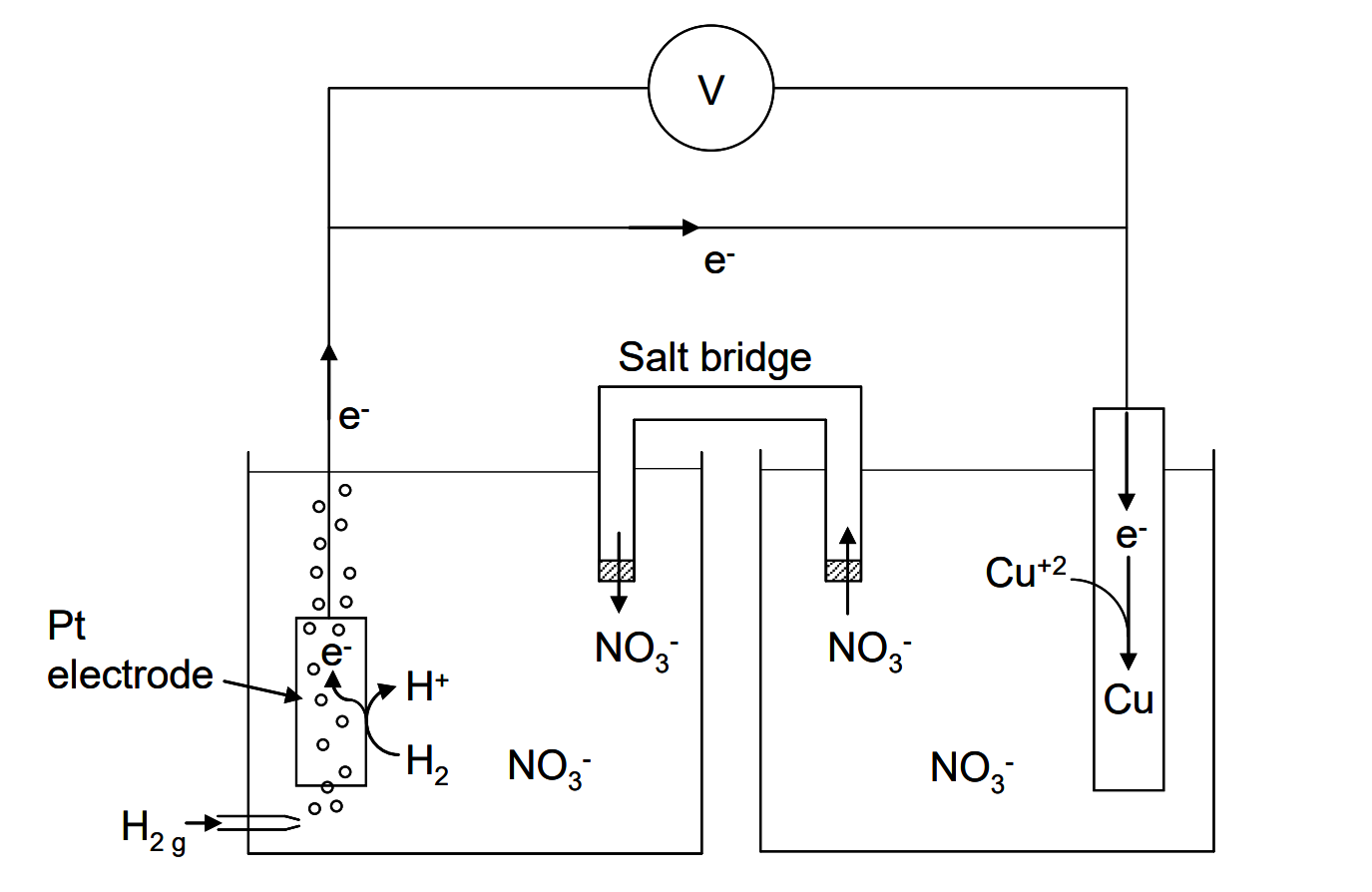
The potential difference is the net effect of two half reactions. There is no way to make a half reaction occur in isolation, and so no way to measure the absolute reduction potential of a half reaction.
The solution to this conundrum is to select one half reaction and assign it a voltage of 0 V. Then all other half reactions are referenced to this one standard. For half reaction (4) then, occurring in a cell as illustrated in Figure 2.1, we measure the potential difference of the cell under some set of standard conditions and arbitrarily say that the half cell voltage for the H+/H2 half reaction will be 0 V. Then the whole of the cell voltage is ascribed to the Cu+2/Cu half reaction. Now we have a measure of the tendency of Cu+2 to undergo reduction relative to the H+/H2 half reaction. Then any half reaction, under this same standard set of conditions (pressure, temperature and composition) can be combined with the H+/H2 half cell and the cell's voltage is then assigned wholly to the half reaction of interest. In other words, every half reaction voltage is precisely equal to the voltage for a cell where the reduction half cell is the half reaction of interest and the anode is the H+/H2 half cell under specified standard conditions.
So, when we set up a cell like that in Figure 2.1, at 25°C and 1 atm pressure with unit activities of the solutes (on the molal scale), we should be able to measure, in principle, a voltage of +0.34 V; the potential difference measured between the cathode (Cu+2/Cu) and the anode (H+/H2), i.e. cathode minus anode*. This is the standard reduction potential for the Cu+2/Cu couple, i.e. half reaction (1). This tells us that Cu+2 has a greater tendency to be reduced than H+by 0.34 V under these conditions. Alternatively, if we were to use the Cr+3/Cu+2 couple instead of Cu+2/Cu in Figure 2.1, we would find that the cell's potential difference would be
-0.42 V (cathode voltage - H+/H2 anode voltage). This indicates that Cr+3 is a weaker oxidizing agent (has a lower potential to be reduced) than H+, or in other words, H+ is 0.42 V more strongly oxidizing than Cr+3.
2.3 Calculating ΔE° for Electron Transfer Reactions
The standard cell voltage is denoted ΔE°, while the standard reduction potential is E°. Under non-standard conditions the superscript ° is dropped.
This may be quite difficult in practice. There are many complicating factors that can make accurate potential measurements difficult or sometimes impossible. However, there are also plenty of indirect methods for obtaining such data. For instance, other thermodynamic measurements may allow us to obtain ΔG° for a reaction. Then through the relationships,
\[\ce{\Delta G^{\circ} = -nF\Delta E^{\circ} = -RT\ln K}\]
the cell voltage can be obtained. Here n is the moles of electrons per mole of reaction and F is the Faraday constant (the charge of a mole of electrons, 96485 C/mole of electrons). These quantities will be developed later.
Most generally we are interested in all electron transfer reactions, not just those that involve the H+/H2 couple. There is a very simple rule for determining the standard cell voltage for any electron transfer reaction:
\[\ce{\Delta E^{\circ} = E^{\circ}_{red} - E^{\circ}_{ox}} \tag{5}\]
where E°red is the standard reduction potential for the couple undergoing reduction, and E°ox is the standard reduction potential for the couple undergoing oxidation. DO NOT change the sign of E°ox. Simply calculate the appropriate difference in standard reduction potentials.
Consider the reaction,
\[
\begin{align*}
&\ce{Cu^{2+}_{aq}}+\ce{2e-}=\ce{Cu_s}\tag{6} \\
&\ce{Fe_s}=\ce{Fe^{2+}_{aq}}+\ce{2e-}\tag{7} \\
\hline
\text{net: }&\ce{Cu^{2+}_{aq}}+\ce{Fe_s}=\ce{Cu_s}+\ce{Fe^{2+}_{aq}}\tag{8}
\end{align*}
\]
(This used to be the basis for recovery of copper metal from dilute copper leach solutions using scrap iron.) The standard reduction potential for Cu+2/Cu is
+0.34 V. That for Fe+2/Fe, i.e. Fe+2 + 2e- = Fe, is -0.44 V. In the reaction Cu+2 is reduced and Fe is oxidized. By equation [1] the standard cell voltage is:
\[\ce{\Delta E^{\circ} = 0.34 - (-0.44) = +0.78\ V} \tag{9}\]
The cell is shown in Figure 2.2. (The two half cells must be separated in order to be able to have the electrons pass through the external circuit and facilitate potential difference measurements, as explained in the figure caption.) So why don't we reverse the sign of E°Fe+2/Fe in order to calculate ΔE°? (Students often want to do this because it seems intuitively reasonable, since the half reaction has been reversed to become an oxidation half reaction.) BUT, the electrons can, in principle, be taken by Cu+2 from Fe, or they can be taken by Fe+2 from Cu. In other words, we have to compare the standard reduction potentials. Hence, do not flip the sign of E°ox. What DE° tells us is the net driving force for the electrons to be accepted by one couple, rather than the other, for the reaction as written. Note that E°Cu+2/Cu > E°Fe+2/Fe. This means that the Cu+2/Cu electrode is positive with respect to the Fe+2/Fe electrode, and then the Fe+2/Fe electrode is negative with respect to the Cu+2/Cu electrode.
If we wrote the reaction as,
\[\ce{Cu_s + Fe^{2+}_{aq} = Cu^{2+}_{aq} + Fe_s} \tag{10}\]
then ΔE° would be,
\[\ce{-0.44 - 0.34 = -0.78\ V} \tag{11}\]
In this case we would be measuring the potential difference of the Fe+2/Fe electrode with respect to the Cu+2/Cu electrode. Now Fe+2 + e- = Fe is the reduction and the oxidation is Cu = Cu+2+ 2e-. Clearly it is important which way round we write the reaction. *
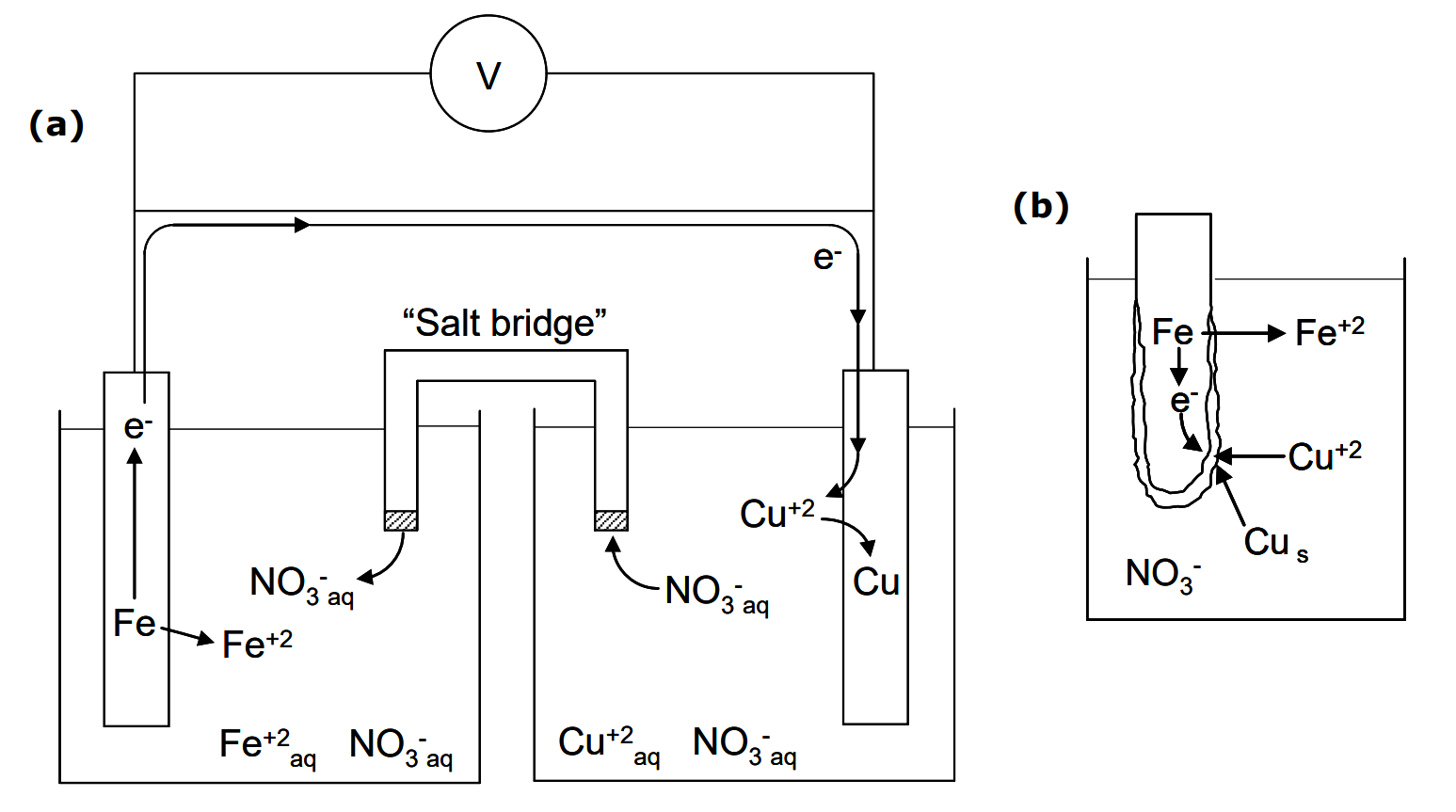
The second common error is to multiply ΔE° by the number of electrons involved (the value of n). The quantity n is the moles of electrons transferred per mole of reaction as written. For reaction (7) n = 2 moles e-/mole of reaction. One mole of reaction as per reaction (7) means 1 mole Cu+2 reacting with 1 mole Fe to form 1 mol Fe+2 and 1 mole Cu. If we wrote the reaction instead as,
\[
\begin{align*}
&\tfrac{1}{2}\ce{Cu^{2+}_{aq}}+\ce{e-}=\tfrac{1}{2}\ce{Cu_s}\tag{12}\\
&\tfrac{1}{2}\ce{Fe_s}=\tfrac{1}{2}\ce{Fe^{2+}_{aq}}+\ce{e-}\tag{13}\\
\hline
\text{net: }&\tfrac{1}{2}\ce{Cu^{2+}_{aq}}+\tfrac{1}{2}\ce{Fe_s}=\tfrac{1}{2}\ce{Cu_s}+\tfrac{1}{2}\ce{Fe^{2+}_{aq}}\tag{14}
\end{align*}
\]
Now 1 mole of reaction means 0.5 mole Cu+2 reacting with 0.5 mole Fe, etc., and n = 1. Multiplying ΔE° by n yields something that is no longer a voltage, but rather,
\[\ce{n\Delta E^{\circ} = moles\ e^- \ volts/mol\ of\ reaction} \tag{15}\]
This is actually more akin to energy. (i.e., converting moles of electrons to charge, we get charge/mole x voltage = energy in J/mole.) Further, the voltage is the potential for electrons to pass from one reactant to another under specified conditions, regardless of the number involved. It is the same if we pass 1 kmol of electrons or 1/1000 mole of electrons. Finally, if we wrote,
\[\ce{10{,}000Cu^{2+}_{aq} + 10{,}000Fe_s = 10{,}000Cu_s + 10{,}000Fe^{2+}_{aq}} \tag{16}\]
(and there is no reason why we can't) would it seem reasonable to multiply 0.78 V by 10,000 and anticipate a cell voltage of 7800 V? Therefore, when calculating a standard potential difference DO NOT multiply ΔE° by n.
2.4 More on E° and ΔE°
We noted earlier that E°, the standard reduction potential, is a measure of the tendency of a species to be reduced. The highest standard reduction potential is about +3 V (e.g. E°F2/F- = 2.87 V). The lowest is about -3 V (e.g. E°Li+/Li = -3.05 V). Thus E°H+/H2 = 0 V is in the middle of the range, which is consistent with the electronegativity for H being about half way between the highest and lowest values. A table of standard reduction (Table 2.1 below) potentials at 25°C is provided below. (Conversions to other temperatures can be performed.) The magnitude of the reduction potential indicates how strongly oxidizing a species is. The more strongly a chemical species wants electrons, the more weakly the reduced form will want to give them up. That is, a strong oxidant is reduced to form a weak reductant. For example, F2 is a very powerful oxidizing agent. It has a high reduction potential (2.87 V). This means that F- has a very weak tendency to give up an electron; it is a very weak reducing agent. Conversely, Li+ is a very weak oxidizing agent; it has a very low reduction potential. Hence Li+ does not have much tendency to accept electrons. It follows then that Li metal is a very powerful reducing agent. The trends are illustrated in the figure below.
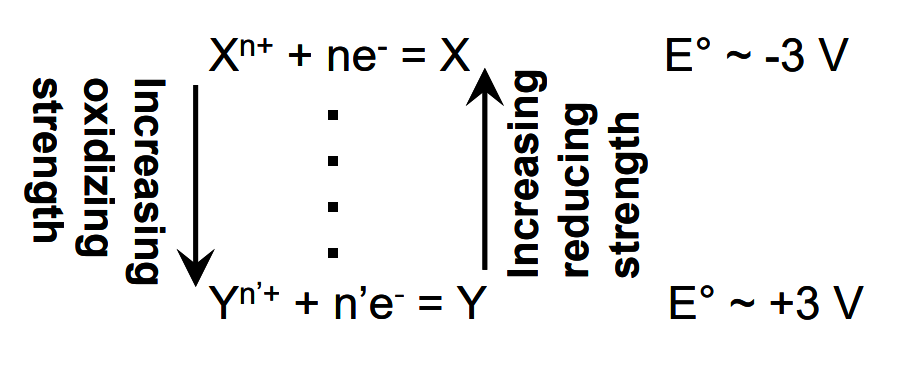
| Table 2.1 - Selected standard reduction potentials in aqueous solutions. The number at the right of each half reaction is its standard reduction potential in volts at 25°C. | |||
|---|---|---|---|
| Sm2+ + 2e- → Sm | - 3.12 | In+ + e- → In | -0.14 |
| Li+ + e - → Li | -3.05 | Pb2+ + 2e- → Pb | -0.13 |
| K+ + e- → K | -2.93 | O2 + H2O + 2e- | -0.08 |
| Rb+ + e- → Rb | -2.92 | HO2- + OH_ | |
| Cs+ + e- → Cs | -2.92 | Fe3+ + 3e- → Fe | -4.40 |
| Ra2+ + 2e- → Ra | -2.92 | Ti4- + e- → Ti3+ | 0.00 |
| Ba2+ + 2e- → Ba | -2.91 | 2H+ + 2e- → H2 | 0 |
| Sr2- + 2e- → Sr | -2.89 | AgBr + e- → Ag + Br- | 0.07 |
| Ca2+ + 2e → Ca | -2.87 | Sn4+ + 2e- → Sn2+ | 0.15 |
| Na+ + e- → Na | -2.71 | Cu2- + e- → Cu+ | 0.16 |
| La3+ + 3e- → La | -2.52 | Bi3 + 3e → Bi | 0.20 |
| Ce3+ + 3e- → Ce | -2.48 | AgCl + e- → Ag + CI- | 0.2223 |
| Mf2+ + 2e- → Mg | -2.36 | Hg2Cl2 + 2e- → 2Hg + 2Cl- | 0.27 |
| Be2+ + 2e_ → Be | -1.85 | Cu2+ + 2e- → Cu | 0.34 |
| U3+ + 3e- → U | -1.79 | O2 + 2H2O + 4e- → 4OH- | 0.40 |
| Al3+ + 3e- → Al | -1.66 | NiOOH + H2O + e- → Ni(OH)2 + OH- | 0.49 |
| Ti2+ + 2e- → Ti | -1.63 | Cu+ + e- → Cu | 0.52 |
| V2+ + 2e- → V | -1.19 | I3- + 2e → 3I- | 0.53 |
| Mn2+ + 2e- → Mn | -1.18 | I2 + 2e- → 2I- | 0.54 |
| Cr2 + 2e- → Cr | -0.91 | Hg2SO4 + 2e- → 2Hg + SO42- | 0.62 |
| Fe(OH)2 + 2e- → Fe + 2OH- | -0.88 | Fe3+ + e- → Fe2+ | 0.77 |
| 2H2O + 2e- → H2 + 2OH- | -0.83 | AgF + e- → Ag + F- | 0.78 |
| Cd(OH)2 + 2e- → Cd + 2OH- | -0.81 | Hg22+ + 2e- → 2Hg | 0.79 |
| Zn2+ + 2e- Ni2+ + 2e- →Ni Zn | -0.76 | Ag- + e- → Ag | 0.80 |
| Cr3- + 3e- → Cr | -0.74 | 2Hg2+ + 2e- Hgz2+ | 0.92 |
| U4+ + e- → U3+ | -0.61 | Pu4+ + e- → Pu3+ | 0.97 |
| O2 - e- → O2- | -0.56 | Br2 + 2e- → 2Br- | 1.09 |
| In3+ s- → In2+ | -0.49 | Pt2+ + 2e- → Pt | 1.20 |
| S + 2e- → S2 | -0.48 | MnO2 + 4H+ + 2e- → Mn2+ + 2H2O | 1.23 |
| In3+ + 2e- → In+ | -0.44 | O2 + 4H+ 4e- → 2H2O | 1.23 |
| Fe2- + 2e → Fe | -0.44 | CrO2O72- + 14H+ + 6e- → 2Cr3+ + 7H2O | 1.33 |
| Cr3- + e- → Cr2+ | -0.41 | Cl2 + 2e- → 2C1- | 1.36 |
| Cd2+ + 2e- → Cd | -0.40 | Au3+ + 3e- → Au | 1.40 |
| In2+ + e- → In+ | -0.40 | Mn3+ + e- → Mn2+ | 1.51 |
| Ti3+ + e- → Ti2+ | -0.37 | MnP4- + 8H+ + 5e → Mn2+ + 4H2O | 1.51 |
| PcSO4 + 2e- → Pb + SO42- | -0.36 | Ce4+ + e- → Ce3+ | 1.61 |
| In3+ + 3e- → In | -0.34 | Pb4+ + 2e- → Pb2+ | 1.67 |
| Co2+ + 2e- → CO | -0.28 | Au+ + e- → Au | 1.69 |
| V3+ + e- → V2+ | -0.26 | Co3+ + e- Ni2+ + 2e- →Ni Co2+ | 1.81 |
| Ni2+ + 2e- →Ni | -0.23 | Ag2+ + e- → Ag+ | 1.98 |
| Agl + e → Ag + I- | -0.15 | 52O82- → 2e- → 28O42- | 2.05 |
| Sn2 + 2e- → Sn | -0.14 | F2 + 2e- → 2F- | 2.87 |
| Source: P.W. Atkins, Physical Chemistry, 3rd ed., 1986. | |||
Standard conditions provides a common basis for comparison of E° values and hence ΔE°. However, there is deeper thermodynamic significance as well. In thermodynamics ΔG (under any conditions, not just standard conditions) is the quantity we use to assess if a reaction at constant pressure is spontaneous or not. It turns out that DE° is directly proportional to ΔG°:
\[\ce{\Delta G^{\circ} = -nF\Delta E^{\circ}} \tag{17}\]
where, as before, n = moles of electrons per mole of reaction and F = the charge of a mole of electrons (96,485 C/mole e-). The units of ΔG° are J/mol. It can be readily seen that on the basis of the units, the equation is reasonable:
In fact, ΔE° provides us with the same information as ΔG°, just on a different scale, and specifically for electron transfer reactions. Based on equation [5] ΔE° > 0 means that a reaction is favourable under standard conditions. Further, the larger the value of DE° the stronger the driving force for the reaction to occur, and the more energy that will be released as a result. Similarly, if DE° < 0 the reaction is not favourable under standard conditions. Making it go will require the input of electrical energy. This is an electrolysis.
2.5 E and ΔE Under Non-Standard Conditions: the Nernst Equation
As noted above, the most general condition for assessing if a reaction as written is spontaneous is if ΔG < 0 under the specified conditions. Changes in conditions can make a reaction favourable or unfavourable, depending on factors such as temperature, pressure and composition. (Note that partial pressure of a gas is analogous to concentration or activity of a solute in solution, or of a metal in an alloy, etc.) Just as ΔG° = -nFΔE°, so,
\[\ce{\Delta G = -nF\Delta E} \tag{19}\]
For a chemical reaction ΔG is a function of pressure, temperature and composition:
\[\ce{\Delta G = \Delta G^{\circ} + RT\ln Q} \tag{20}\]
where R is the gas constant (8.31441 J/mol K), T is the absolute temperature (Kelvin) and Q is the reaction quotient. The reaction quotient was presented in Chemistry Review Part II.
By definition 1 VC = 1 J. When a charge of 1 C passes through a voltage of 1 V an energy of 1 J is consumed or released, depending on whether the charge is going from + to – or from – to +. Introductory textbooks on the subject typically show that,
\[\ce{-w_e = \Delta G^{\circ} = -nF\Delta E^{\circ}}\]
where -we is the maximum possible electrical work (or more generally, non pressure-volume work) that can be extracted from an electrochemical cell.
Briefly, it has exactly the same form as the equilibrium constant, but where concentrations and/or gas pressures (activities and fugacities, properly speaking) are not necessarily those corresponding to an equilibrium state. Q is an indicator of how far the system is from equilibrium. The reaction will naturally proceed with changes in composition until equilibrium is attained, i.e. Q → K. In order to force Q to diverge from K, energy must be supplied (e.g. an electrolysis). Another important relationship is,
\[\ce{\Delta G^{\circ} = -RT\ln K = -nF\Delta E^{\circ}} \tag{21}\]
(This is another important aspect of standard conditions.) Note that when ΔG = 0, ΔG° = -RTln Q (equation [8]) and now Q = K. Thus all reactions naturally tend to drive towards ΔG going to 0. Once this condition is reached, Q = K and the reaction has reached equilibrium. There will be no further net change in composition. Substituting ΔG = -nFΔE into equation [8] yields,
\[\ce{-nF\Delta E = -nF\Delta E^{\circ} + RT\ln Q} \tag{22}\]
\[\ce{\Delta E = \Delta E^{\circ} - \frac{RT}{nF}\ln Q} \tag{23}\]
This is the Nernst equation. For a spontaneous electron transfer reaction, ΔE is a measure of how far the reaction is away from equilibrium. All natural processes tend toward decreasing ΔE. Once equilibrium is attained ΔE goes to 0 and the reaction ceases. For a galvanic cell (a battery), the voltage declines with time and eventually the battery dies. If ΔE < 0 then the reaction as written is unfavourable. To make it go in this direction will require energy to overcome the negative ΔE.
By this point it can be seen that if the reaction is reversed, so is the sign of ΔG (or ΔG°); if it is favourable in the direction written (ΔG < 0), then it is unfavourable in the opposite direction (and ΔG > 0). Likewise, reversing the direction of an electron transfer reaction requires that we reverse the sign of ΔE° (or ΔE for non-standard conditions).
2.6 The Standard Oxidation Potential
Sometimes people characterize half reactions by standard oxidation potentials, rather than standard reduction potentials. This can be confusing. As always in thermodynamics, keeping the conventions straight is crucially important. A generic oxidation half reaction is,
\[\ce{M_s = M^{n+} + ne^-} \tag{24}\]
Consider again the Cu+2/Cu reduction half reaction, Cu+2 + 2e- = Cu. We defined the standard reduction potential as the cell voltage (cathode - anode) where the cathode was the Cu+2/Cu couple and the anode was the H+/H2 couple (set to 0 V). It tells us that Cu+2 is 0.34 V more prone to accept electrons than is H+. And if you set up the cell, as in Figure 2.1, and measure the potential of the Cu+2/Cu electrode with respect to the H+/H2 electrode, the voltage will be +0.34 V. This means that Cu is more prone to hold onto its electrons than is H2; Cu is harder to oxidize than H2. Quantitatively, H2 is 0.34 V more prone to be oxidized than Cu, or, since E°H+/H2 = 0 V (by convention, for either its reduction or oxidation potential), the oxidation potential for the Cu+2/Cu couple is -0.34 V with respect to standard H+/H2. In other words, the standard oxidation potential is just the negative of the standard reduction potential.
Regardless, the cell voltages work out the same. For the moment designate the standard reduction potential as E°red and the standard oxidation potential as E°ox. Consider again the reaction,
\[\ce{Cu^{2+}_{aq} + Fe_s = Cu_s + Fe^{2+}_{aq}} \tag{25}\]
From the perspective of reduction potentials,
\[\ce{Cu^{2+}_{aq} + 2e^- = Cu_s}\qquad E^{\circ}_{red} = +0.34\ V \tag{26}\]
\[\ce{Fe^{2+}_{aq} + 2e^- = Fe_s}\qquad E^{\circ}_{red} = -0.44\ V \tag{27}\]
\[\ce{\Delta E^{\circ} = E^{\circ}_{red} - E^{\circ}_{ox}
= E^{\circ}_{red,cathode} - E^{\circ}_{red,anode}
= E^{\circ}_{red,Cu^{2+}/Cu} - E^{\circ}_{red,Fe^{2+}/Fe}}\]
\[\ce{= 0.34 - (-0.44) = +0.78\ V} \tag{28}\]
From the point of view of oxidation potentials,
\[\ce{Cu_s = Cu^{2+}_{aq} + 2e^-}\qquad E^{\circ}_{ox} = -0.34\ V \tag{29}\]
\[\ce{Fe_s = Fe^{2+}_{aq} + 2e^-}\qquad E^{\circ}_{ox} = +0.44\ V \tag{30}\]
Analogously to the preceding case,
\[\ce{\Delta E^{\circ}
= E^{\circ}_{ox} - E^{\circ}_{red}
= E^{\circ}_{ox,anode} - E^{\circ}_{ox,cathode}
= E^{\circ}_{Fe/Fe^{2+}} - E^{\circ}_{Cu/Cu^{2+}}
= 0.44 - (-0.34) = +0.78\ V} \tag{31}\]
In this text we will only use reduction potentials.
Media Attributions
- Ch3_F1_H-Cu_Galvanic_Cell © Bé Wassink and Amir M. Dehkoda is licensed under a CC BY-NC (Attribution NonCommercial) license
- Ch3_F2_Cu-Fe_Galvanic_Cell © Bé Wassink and Amir M. Dehkoda is licensed under a CC BY-NC (Attribution NonCommercial) license
- Ch3_F3_Redox_Strength_Trend © Bé Wassink and Amir M. Dehkoda is licensed under a CC BY-NC (Attribution NonCommercial) license
One of several types of apparatus for joining half cells that allows for electrical connection between them. A salt bridge is usually comprised of a U-shaped tube filled with an aqueous gel, such as agar, that contains a high concentration of a simple salt, like KCl or KNO3. The salt is chosen so as to be compatible with both half cells; the salt must not interfere with the half reactions. Electrons flowing through the external circuit of a cell establish a current. For the circuit to be complete there must be provision for the current to pass through the solution. Electrons can't do that; they do not survive in solution. Rather the current is carried by migration of ions in solution. Anions flow out of the reduction half cell and into the anode half cell. Equivalently, cations can move out of the anode half cell and into the cathode half cell. This maintains electrical neutrality. Without this the current flow would cease virtually immediately. (Bulk charge separation is enormously energetic, as a lightning storm amply demonstrates.) Other methods can also be used to facilitate ionic migration, such as ion exchange membranes.
An electrode where oxidation occurs.
An electrode where reduction occurs.
