Chapter VII: Electrowinning
3. Thermodynamics of Electrochemical Cells
Reversible and Irreversible Processes
In thermodynamics a reversible process is one for which the direction of a process (such as a reaction) can be reversed by an infinitesimal change. For example, if a process is operating reversibly, an infinitesimal change in pressure or temperature or concentration can reverse the direction of the process. Reversible processes are in thermal equilibrium with their surroundings. They are also at equilibrium in other respects, e.g. chemically or mechanically. How then can there be any actual change of state? Suppose there is a chemical reaction occurring, A = B. And suppose the system is at equilibrium. Increase the concentration of A by d[A], an infinitesimal change. The reaction proceeds to the right to an infinitesimal degree. Continue to increase the concentration of A in infinitesimal steps. The reaction proceeds to produce additional concentration of B by d[B] increments. In the limit of infinite time a finite extent of reaction will have occurred. Note that at any stage during the process the reaction can be reversed by adding an infinitesimal concentration of B. This is a reversible process. Truly reversible processes are of no practical use; they occur infinitely slowly. But, they are a condition or case that thermodynamics can use to tell us something about theoretical limiting possibilities. It helps us to answer questions like, "What is the minimum possible heat we can put into a process to make it go?" Or, "What is the maximum possible work we can get out of a process?"
Naturally then, real processes are always irreversible. (Irreversible does not mean that it cannot be reversed, but, rather the opposite of thermodynamically reversible.) They have a finite (not infinitesimal) driving force to proceed in one direction. Stopping or reversing the process requires a finite change in a variable. Real processes sometimes can approach, but never truly attain reversibility. A reversible process always has associated with it the minimum possible heat flow. (This can be qualitatively understood from an example. If you very slowly and gently set down a large rock on a surface there will be little or no perceptible change in temperature of the rock and the surface. If you drop the rock it will hit the floor with substantial force and generate a substantial rise in temperature. Both cases involved the same change in gravitational potential energy. The latter was the most irreversible case.)
Real processes involve conditions that are far from equilibrium. They move spontaneously towards equilibrium. If a process is exothermic, for instance, the heat loss is larger than would be the case under reversible conditions. Heat loss from the system is negative and qirrev < qrev, i.e. qirrev is a bigger negative number than qrev (|qirrev| > |qrev|).
With respect to galvanic electrochemical cells (favourable reaction) under hypothetical reversible conditions, the heat flow is the minimum, while the work that can be done is the maximum. Recall that the change in internal energy for a change of state (e.g. 1 mol Aaq → 1 mol Baq, as above) is the sum of heat flow minus work flow, (ΔU = q - w; work done by the system is positive by definition and heat exiting the system is negative.) Internal energy is a state function, i.e. it depends only on the final and initial states, not how you get from one to the other. Then, the less heat evolved for a given change, the more work that was extracted from that change. For a real cell, operated under real conditions, the process is necessarily irreversible and the heat flow is greater than in the reversible case, so the work obtainable is less. The farther from reversibility (or the more irreversible the process), the more the heat and the less the obtainable work. Extending the idea that reversible processes run infinitely slowly, the faster the process is run, i.e. the more rapidly the battery is discharged, the more irreversible the process, and the more of the energy that is lost as heat.) The reversible case defines the limiting possibility.
Some examples of irreversible processes include:
- Flow of heat from a hot body to a cold one
- Water flowing downhill
- Hydrometallurgical leaching reactions
- The conversion of chemical energy into electrical energy in galvanic cells.
- An electrolysis.
Once the final equilibrium state is reached the capacity of the system to do further work is exhausted. In example 1, the two bodies reach the same temperature, and in example 4 the battery goes dead. Real processes involve finite changes of state with finite energy changes. There is a driving force, or potential for the process to occur. If the change is thermodynamically favourable then the process is spontaneous and irreversible. (Recall that for a spontaneous process, ΔG < 0. The Gibbs free energy function expresses the requirement that spontaneous processes must increase the net entropy of the system plus its surroundings.) If the process is not favourable it is not spontaneous and does not naturally tend to occur. The reverse (opposite) process is actually favoured and naturally does tend to occur. The non-spontaneous process can be forced to occur by input of sufficient energy, and when this is done the real process is also irreversible.
A Review of Some Relevant Thermodynamics
Next, a refresher on some aspects of the thermodynamics related to electrochemical cells is needed. Enthalpy is the sum of internal energy + PV, where P = pressure and V = volume.
\[H = U + P V \tag{37}\]
U = q – w, staying with the engineering convention that work done by the system is positive and heat flow out of the system is negative.
\[dH = dq - dw + d(PV) \tag{38}\]
(H is very similar to U, but more convenient at constant pressure.)
For a reversible process the heat flow is denoted dqrev. Then,
\[dq_{\mathrm{rev}} = T\,dS \tag{39}\]
where T is the absolute temperature and dS is the entropy* change of the system (this from the definition of entropy). Under reversible conditions, the system can do its maximum possible work, and the heat flow is the minimum possible, i.e.
\[-dw = -dw_{\mathrm{rev}} = \text{maximum work possible} \tag{40}\]
\[G = H - T S \tag{41}\]
where G is the Gibbs free energy. At constant temperature, reversible conditions:
\[dG = dH - T dS \tag{42}\]
\[dG = dU + d(PV) - T dS \tag{43}\]
\[dG = dq_{\mathrm{rev}} - dw_{\mathrm{rev}} + d(PV) - T dS \tag{44}\]
\[dG = T dS - dw_{\mathrm{rev}} + d(PV) - T dS \tag{45}\]
Generally, the work is comprised of pressure-volume work and non-PV work, such as electrical, gravitational etc. (the former is of interest here). The work term is,
\[-dw_{\mathrm{rev}} = -P\,dV - dw'_{\mathrm{rev}} \tag{46}\]
where dw’rev is the non-PV work (electrical work here) under reversible conditions. At constant pressure,
\[dG = -P\,dV - dw'_{\mathrm{rev}} + P\,dV + V\,dP = -dw'_{\mathrm{rev}} \tag{47}\]
\[\Delta G = -w'_{\mathrm{rev}} \tag{48}\]
since dP = 0 (constant pressure). (The cancelling of the PdV terms is what makes the enthalpy function convenient.) Hence the maximum non-PV work (w’rev) is equal to -ΔG (at fixed P, T), and this is obtainable only under reversible conditions. This is a limiting case. Electrochemical cells commonly do operate under conditions of constant temperature and pressure. However, real cells cannot operate under truly reversible conditions. Sometimes real cells may come moderately close.
Entropy can be thought of as the inverse of the "concentration" or "quality" of energy. Energy naturally tends to disperse: heat flows to cooler bodies, unequal concentrations tend to equalize, light moves away from its source, and so on. All this occurs naturally without having to be forced. It just happens. Thus the "concentration" of energy always tends to drop; energy wants to become more diffuse. This is the entropy effect. Thermodynamically speaking, entropy naturally tends to increase; the dispersal of energy increases. THIS IS WHAT DRIVES ALL SPONTANEOUS PROCESSES. If a process is spontaneous (or favourable) it means that overall there is a net increase in entropy; a degradation of the "concentration" of energy. It may occur within the system of interest (a reaction in a cell, for instance) or it may occur in the environment surrounding the system (the "surroundings") or both. Regardless, it is the inviolable requirement for any process to be spontaneous.
What is so marvelous about the Gibbs free energy function is that it accounts for both the change in entropy in the system and in its surroundings. To provide a brief and less than rigorous rationale for this, consider that ΔG = ΔH - (TS) = H - TΔS at constant temperature. Then ΔG/T = ΔH/T - ΔS, where ΔS is the entropy change for the system. And, ΔH/T is q/T when the pressure is constant (U = q - w and at constant pressure w = PΔV, i.e. pressure-volume work, such as the expansion of a gas. Then ΔH = q - PΔV + PΔV = q = heat flow at constant pressure, often denoted qP.) Under reversible conditions ΔH/T = qrev/T = the heat flow into the surroundings over T. Then ΔH/T is the entropy change in the surroundings. The minus sign in ΔG = ΔH - TΔS accounts for the fact that heat flow out of the system into the surroundings is the opposite of heat flow in the surroundings to the system; it takes care of the sign convention issues. Thus if ΔG < 0 there is a net increase in entropy within the system + surroundings, and the process is spontaneous. If ΔG > 0 the reaction is unfavourable (not spontaneous); if it were to occur there would be a net decrease in entropy. This cannot naturally occur, though it can be forced with energy input.
Energy relations for electrochemical cells
Recall the first law of thermodynamics, which states “the energy of the universe is constant,” or “energy is neither created nor destroyed.” In any system energy can be transferred to or from the surroundings as heat or work. These are forms of energy in transit, i.e. both are flows of energy. This is expressed mathematically as,
\[\Delta U = q - w \tag{49}\]
The equation follows the engineering sign convention where,
q < 0 means heat flows out of the system into the surroundings (exothermic)
q > 0 means heat flows into system from surroundings (endothermic)
w < 0 means work flows into the system from surroundings
w > 0 means work flows out of system into the surroundings
Heat flow is considered from the perspective of the system, while work flow is considered from the perspective of the surroundings. (In the SI convention both work and heat flows are considered from the perspective of the system. Either means of energy flow into the system is positive; either means of energy flow out of the system is negative. Most chemistry texts follow the latter convention.) By definition,
\[G = U + P V - T S = H - T S \tag{50}\]
If a system undergoes a change from state 1 to state 2,
\[G_2 - G_1 = U_2 - U_1 + (P_2 V_2 - P_1 V_1) - (T_2 S_2 - T_1 S_1) \tag{51}\]
\[U_2 - U_1 = q - w \tag{52}\]
\[G_2 - G_1 = q - w + (P_2 V_2 - P_1 V_1) - (T_2 S_2 - T_1 S_1) \tag{53}\]
and at fixed P and T,
\[G_2 - G_1 = q - w + P\,(V_2 - V_1) - T\,(S_2 - S_1) \tag{54}\]
w is the total work, including work other than pressure-volume work (PV work; e.g. expansion of a gas against some external pressure P). For an electrochemical cell where electrical work is also possible (by means of electrons flowing through an external circuit),
\[w = w' + P\,(V_2 - V_1) \tag{55}\]
where w’ represents the electrochemical work. Substituting this into the preceding equation yields,
\[G_2 - G_1 = q - w' - T\,(S_2 - S_1) \tag{56}\]
or
\[\Delta G = q - w' - T\,\Delta S \tag{57}\]
Again, this applies to a process at fixed P and T. Since,
\[\Delta G = \Delta H - T\,\Delta S \tag{58}\]
It is then apparent that,
\[\Delta H = q - w' \tag{59}\]
which is equal to the heat flow at constant pressure. For a change of state achieved reversibly, at constant temperature and pressure, we would have the equation,
\[\Delta G = q_{\mathrm{rev}} - w'_{\mathrm{rev}} - T\,\Delta S \tag{60}\]
An electrochemical cell for which ΔG < 0 (favourable or spontaneous) operated reversibly will generate the maximum possible amount of electrical work,
\[-w'_{\mathrm{rev}} = -w'_{\max} = \Delta G \tag{61}\]
as per equation {48}. Substituting this into the equation above,
\[\Delta G = q_{\mathrm{rev}} + \Delta G - T\,\Delta S \tag{62}\]
or, as we would expect,
\[q_{\mathrm{rev}} = T\,\Delta S \quad \text{(at constant temperature)} \tag{63}\]
This is the flow of energy as heat in a cell operated reversibly and represents the minimum possible heat loss (again for a cell where ΔG < 0 which does electrical work). Real cells, operated irreversibly will generate less work and more heat. For an electrolytic cell (ΔG > 0; not favourable), the minimum possible work that we can input to force a reaction to go in the unfavourable direction is again -w'rev (< 0). To make the reaction go at a practical rate, making the process irreversible, the actual work input will be > -w'rev.
Definitions
The cell potential is also called the EMF (electromotive force). It is the voltage of a cell under reversible conditions (no current is flowing, or, the current is infinitely small). It represents the driving force for electron transfer. When ΔE > 0 the cell reaction is spontaneous. When ΔE < 0 the reaction as written is not spontaneous, i.e. is not favoured. (Recall that ΔG = -nFΔE). In this discussion, in order to distinguish charge from heat flow, the charge will be symbolized as qc. Work (in joules) = voltage (V) x charge (C). The cell potential ΔE and the reversible electrical work w’rev have the same sign (for the engineering convention, not the SI convention) and are related as follows,
\[-\Delta G = nF\,\Delta E = w'_{\mathrm{rev}} = q_c\,\Delta E \tag{64}\]
Types of Electrochemical Cells
Now different types of electrochemical cells can be compared along with their energy relations. The four common types of cells are illustrated in the Figure 3.1 below. A piece of zinc is suspended in a solution of ZnSO4 and H2SO4. The other electrode is a piece of platinum. Hydrogen gas is bubbled over the platinum surface. The H+/H2 half reaction is rapid on platinum. (Rates of electron transfer depend strongly on the surface at which they occur.) The half reactions are:
\[\ce{2H+ + 2e- -> H2} \qquad E^\circ = 0~\text{V} \tag{65}\]
\[\ce{Zn^{2+} + 2e- -> Zn} \qquad E^\circ = -0.76~\text{V} \tag{66}\]
There are two possibilities. The favourable reaction may occur, for which ΔE > 0. Alternatively the reaction can be forced to go in the opposite direction by applying a suitably high opposing voltage. This is electrolysis. The favourable reaction involves oxidation of Zn to Zn+2 and reduction of H+ to H2. In fact there is no thermodynamic reason why the reaction should not spontaneously occur directly on the zinc surface, as illustrated in Figure 3.2 below. However, the reduction of H+ on very pure Zn is very slow, whereas it is quite rapid on Pt. Because the reduction of H+ on pure Zn is so slow, the cell can be set up with Zn metal in direct contact with H+. Otherwise two half cells with provision for ionic conduction would be used.
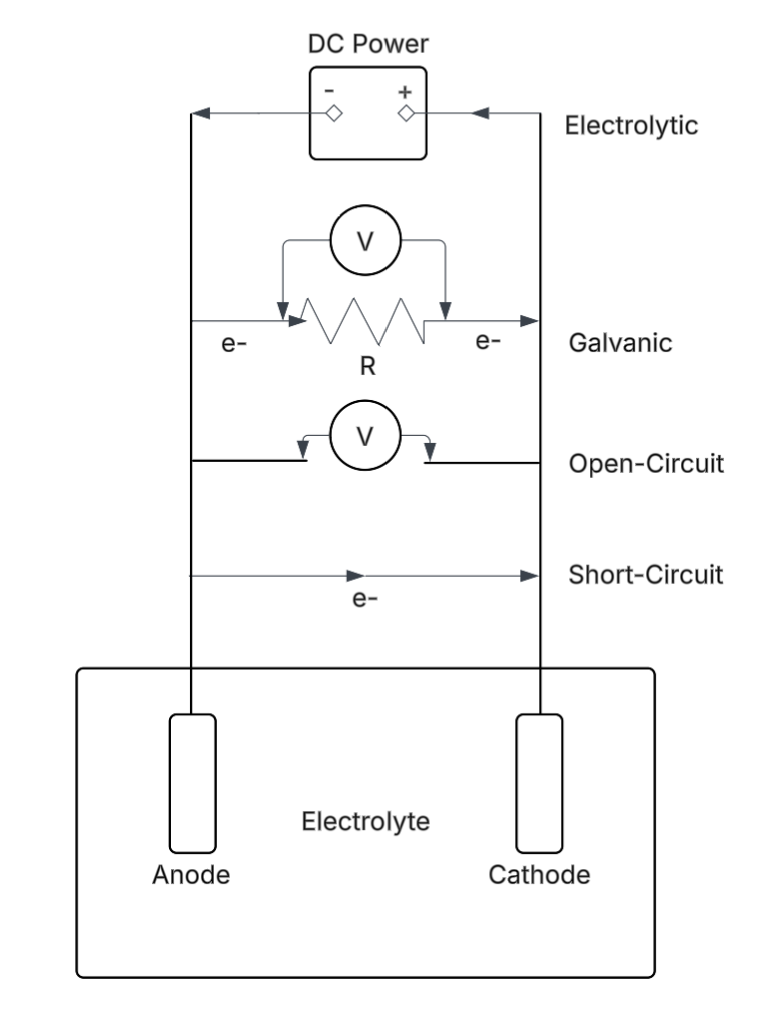
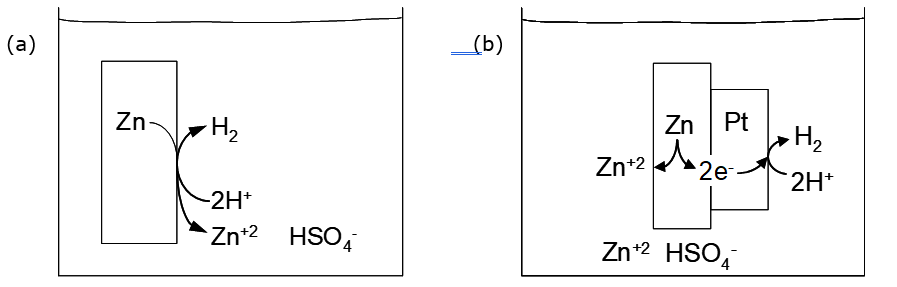
1. Short-circuited cell
There is no load (no electrical work is extracted) in the system. The reaction proceeds spontaneously since ΔE° >0 (or
ΔE > 0 for non-standard conditions) and the process is favourable.
\[\ce{Zn -> Zn^{2+} + 2e-} \qquad E^\circ = -0.76~\text{V} \tag{67}\]
\[\ce{2H+ + 2e- -> H2} \qquad E^\circ = 0~\text{V} \tag{68}\]
The overall reaction is:
\[\ce{Zn + 2H+ -> Zn^{2+} + H2}
\qquad\Delta E^\circ = 0 - (-0.76) = 0.76~\text{V} \tag{69}\]
This is really equivalent to the situation in Figure 3.1 (b). Since the reduction of H+ on pure Zn is slow, all that is required for the reaction to proceed at a substantial rate is a catalyst, which is the role of Pt.
\[\Delta G^\circ= -nF\,\Delta E^\circ= -2~\text{mol e}^{-}\!\!/\text{mol} \times 96485~\text{C/mol e}^{-} \times0.76~\text{V}= -1.47 \times 10^{5}~\text{VC/mol}= -1.47 \times 10^{5}~\text{J/mol}= -147~\text{kJ/mol}\tag{70}\]
Since,
\[\Delta H = q - w' \quad \text{and} \quad w' = 0 \tag{71}\]
(no electrical work is being done; there is no load),
\[\Delta H = q \tag{72}\]
All the energy is dissipated as heat. Heat flows from the system (the cell) to the surroundings, and so by convention is negative. The process is exothermic. (If you have ever short-circuited a battery by connecting a wire across both ends, you know this from experience; the wire can get red hot and the rate of discharge of the cell can get dangerously fast.) Since there is no work being extracted, the potential difference is zero (w’ = 0 = ΔEqc. Therefore ΔE = 0). Note that the cell in principle is capable of manifesting a voltage ΔE, but when short-circuited this is not realized. Cementation reactions and corrosion processes are examples of short-circuited cells. Any redox reaction occurring directly between reagents without running the electron transfer through an external circuit is a short-circuited cell. So is a cell where a wire is connected across the poles. Other examples include oxidative leaching processes of sulfides and combustion reactions.
2. Open-circuit cell
The half reactions are the same as in the short circuited cell. However, if the electrical connection between the two electrodes is broken, no current flows. A voltmeter can be used to measure the potential difference. This may employ a very large resistance inside the meter. Then the current flow is so small that the rate of reaction also is extremely slow. This very nearly approaches the reversible case (a process carried out infinitely slowly). (Alternatively, an opposing voltage can be applied until the current is zero. The opposing voltage slows the reaction until a point where the reaction stops. At this point the opposing voltage is equal to the cell voltage. These devices, called potentiometers, are not much in use anymore.) Under standard conditions (298 K, PH2 = 1 atm, unit activities of the ions) the measured potential difference is 0.76 V. This indicates the thermodynamic potential difference, or driving force for the reaction. If the conditions were non-standard the potential difference would differ from ΔE°, as per the Nernst equation, and this would indicate the driving force under those conditions. In principle, open circuit cells can be used to measure thermodynamic potentials. In practice, it is often not so easy for many reasons.
An open circuit cell is essentially a cell working reversibly; the rate of the reactions is infinitely slow by virtue of the open circuit. The cell voltage equals the thermodynamic potential. The cell can do its maximum possible work. For all practical purposes, however, we can't extract work from such a cell in a finite time. It's of no practical use as far as obtaining electrical work. It is of use for measuring cell potentials. For practical work we use the cell galvanically or electrolytically.
3. Galvanic cell (battery)
In this case the electrodes are connected to a moderately high resistance device that uses electrical energy as work, such as a radio. The half reactions are the same as in the short-circuited cell. The reaction is favourable. The current passes at a fairly low, but, finite rate. This might be close to reversible conditions of operation, though it is irreversible. Hence the heat loss must be somewhat greater than the minimum reversible process heat loss, and the work obtained must be somewhat less than it would be under truly reversible conditions. Since the same charge is being passed as it would be under reversible conditions (2e- per Zn+2) and the available work is less, the cell voltage (denoted V) must be lower:
\[\left( w' = V\,q_c \right) \;<\; \left( w'_{\mathrm{rev}} = \Delta E\, q_c \right) \tag{73}\]
Therefore V < ΔE (ΔE is the cell voltage under reversible conditions; the maximum possible voltage). The greater the current, the faster the process and the greater the extent of departure from reversibility. Then the heat loss is greater and the work that can be extracted is lower. One way to rationalize this is that as the current gets high, the process is getting closer to operating like a short-circuited cell, where all the energy is dissipated as heat.
The thermodynamic potential for the reaction is given by the Nernst equation:
\[\Delta E= \Delta E^\circ- \frac{RT}{nF}\ln\!\left(\frac{a_{\ce{Zn^{2+}}}\, P_{\ce{H2}}}{a_{\ce{H+}}^{2}}\right)\tag{74}\]
Say PH2 is kept constant at 1 atm. As the reaction proceeds [Zn+2] increases, while aH+ decreases. The ln term thus increases as the reaction proceeds and ΔE drops. Eventually equilibrium is reached and no further reaction occurs. Then ΔE goes to zero. ΔE then is a measure of how far away from equilibrium the system is; how great the driving force is for chemical reaction to occur.
The process will continue until chemical equilibrium is reached, at which point ΔG and ΔE for the cell both go to zero. At equilibrium there is no more driving force for the reaction to proceed; no further change occurs. At this point the battery is dead. All real galvanic cells operate at less than the thermodynamic limit of efficiency. However, if the cell is discharged slowly, the efficiency approaches that of a reversibly operated cell. This is why fuel cells are naturally quite efficient. A fuel cell is simply a galvanic cell in which the reactants are continuously replenished, and the products are continuously removed.
The thermodynamics can be conveniently represented on a diagram as shown below.
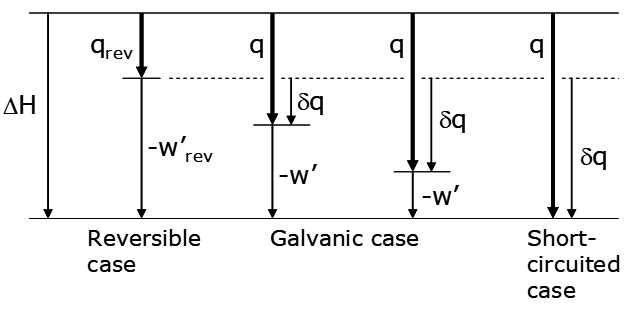
Recall that ΔH is a state function, meaning that for going from a specified initial state (e.g. the left side of reaction (69) at a given temperature, pressure and concentrations) to a specified final state (e.g. the right side of reaction (69) with specified temperature, pressure and concentrations) the change in enthalpy is the same no matter how the change is effected, be it reversibly, galvanically or as a short-circuited cell. In this case ΔH < 0; energy both as heat and work leave the system. What does depend on how we run the cell is q and w', but the sum, q - w', is always the same. The limiting case is the reversible cell; |qrev| is the minimum possible heat flow and wrev is the maximum possible work. Galvanic cells operate at finite rates, are irreversible in the thermodynamic sense and exhibit larger heat losses and lesser capabilities for work; |q| > |qrev| and w' < w'rev. In addition, the faster the cell is operated (the greater the current) the more irreversible it is and the greater the heat flow and the less the work. The quantity Δq is the difference between w'rev and w':
\[q = q_{\mathrm{rev}} + \delta q \tag{75}\]
\[\Delta G = -w'_{\mathrm{rev}} = -nF\,\Delta E \tag{76}\]
Based on equation {54},
\[q = \Delta G + w' + T\,\Delta S \tag{77}\]
\[q = \Delta G + w' + q_{\mathrm{rev}} \tag{78}\]
\[q = -w'_{\mathrm{rev}} + w' + q_{\mathrm{rev}} \tag{79}\]
Then,
\[\delta q = -w'_{\mathrm{rev}} + w' \tag{80}\]
\[-\delta q = w'_{\mathrm{rev}} - w' \tag{81}\]
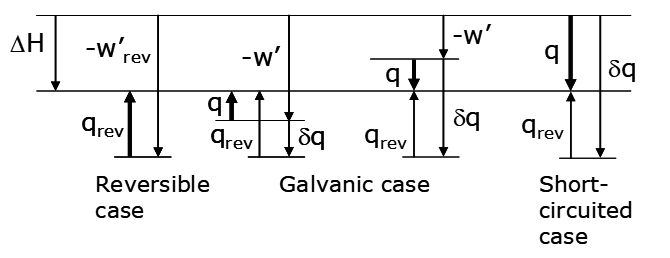
The heat loss is the sum of qrev + δq (which is < 0). Thus the additional heat loss arises from inefficiency in operating the cell, relative to the limiting, reversible case. If the cell is short-circuited no work can be extracted and all the energy output is lost as heat. This is the other limiting case. Another possibility for a galvanic cell is when qrev > 0. This is depicted in the alternative diagram below.
4. Electrolytic cell.
This is what is employed in electrowinning. The electrodes are connected to an external power supply such that the voltage exceeds and opposes the thermodynamic cell voltage. In electrolysis the reaction is being forced in the opposite direction of its natural or spontaneous direction. The half reactions now are,
\[\ce{Zn^{2+} + 2e- -> Zn} \tag{82}\]
\[\ce{H2 -> 2H+ + 2e-} \tag{83}\]
The overall reaction is how the reverse of reaction (69):
\[\ce{Zn^{2+} + H2 -> Zn + 2H+}
\qquad\Delta E^\circ = -0.76~\text{V}\tag{84}\]
This is NOT favourable and will not occur naturally. To overcome this, a voltage of >0.76 V (under standard conditions) is applied externally to force the reaction to go as written above, i.e. ΔEappl > 0.76 V. The flow of electrons is reversed and so are the electrode reactions relative to the galvanic or short-circuited cases. The H2/H+ reaction now becomes the anode and the Zn+2/Zn process becomes the cathode. The reaction will reach equilibrium when the thermodynamic cell voltage reaches ΔEappl. Then it will stop. The thermodynamic potential for the reaction is given by the Nernst equation:
\[\Delta E= \Delta E^\circ- \frac{RT}{nF}\ln\!\left(\frac{a_{\ce{H+}}^{2}}{a_{\ce{Zn^{2+}}}\, P_{\ce{H2}}}\right)\tag{85}\]
Say PH2 is fixed at 1 atm. As the reaction proceeds aH+ increases and aZn+2 decreases. Hence the log term increases and consequently ΔE decreases (becomes a larger negative number) as the reaction proceeds. This will continue until |ΔE| and ΔEappl are equal. Then the thermodynamic cell potential is just balanced by the applied potential and there is no net potential difference between the electrodes. The reaction stops. To make the reaction proceed further still, one would have to increase ΔEappl. Under conditions of fixed external potential, the reaction rate would decrease as the thermodynamic cell voltage decreases (because the driving force, which is the difference between ΔEappl and the thermodynamic |ΔE|, decreases). In practice, electrowinning is carried out under conditions of controlled current, rather than controlled potential, as was explained in the section on Faraday's Law relationships. As reactants are depleted, the applied voltage must increase to maintain the constant current. In practice, electrowinning usually takes less than 50% of the desired metal ion from the solution. The barren electrolyte after EW is recycled to increase the metal ion tenor. Considering that ΔE changes by,
\[\frac{2.303\,RT}{2F}\log\!\left(\frac{P_{\ce{H2}}\, a_{\ce{Zn^{2+}}}}{a_{\ce{H+}}^{2}}\right)
=
0.02958\,\log\!\left(\frac{P_{\ce{H2}}\, a_{\ce{Zn^{2+}}}}{a_{\ce{H+}}^{2}}\right)\tag{86}\]
even a 50% change in concentrations has only a small effect on the cell voltage.
Now work is being done on the cell by the surroundings; an external voltage is applied. Then w’ < 0, and the work done on the cell is given by,
\[w' = -nF\,\Delta E_{\mathrm{appl}} \tag{87}\]
where ΔEappl is a positive number. The work done on the cell is directly proportional to the applied voltage. The energy changes are summarized in the diagrams below.
\[\Delta H = q - w' \tag{88}\]
Note that w' < 0, w’ < w’rev and -w' > -w'rev. The minimum work required to drive the reaction against its favourable direction (backwards) to effect electrolysis is -w'rev. In practice more electrical work is required to obtain reasonable rates; -w' > -w'rev.
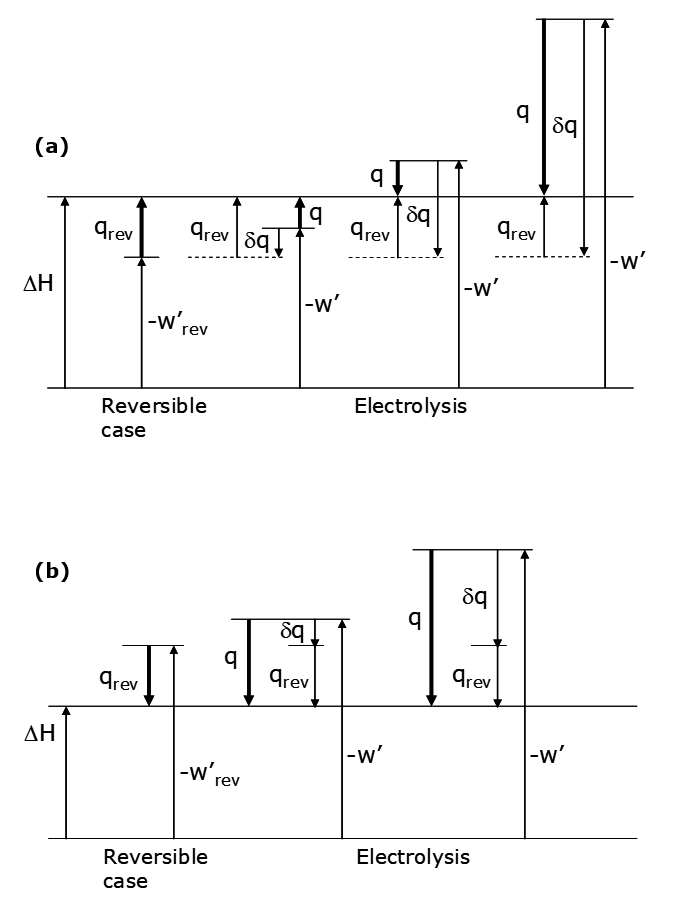
The net heat is the sum:
\[q = q_{\mathrm{rev}} + \delta q= q_{\mathrm{rev}} - w'_{\mathrm{rev}} + w'\tag{89}\]
If qrev > 0, the sign of q depends on the magnitude of w', which in turn depends on the magnitude of ΔEappl (Figure 3.4 a). Once ΔEappl gets large enough there is a net heat flow from the cell into the surroundings. Practical electrowinning usually uses ΔEappl >> -ΔE (the thermodynamic cell voltage) so that heat will be evolved. If qrev < 0 the sign of q is always negative, as indicated in Figure 3.4 b. Referring again to the equation,
\[q = \Delta G + w' + T\,\Delta S \tag{90}\]
Substituting in,
\[w' = -nF\,\Delta E_{\mathrm{appl}}\qquad\text{and}\qquadw'_{\mathrm{rev}} = -nF\,\Delta E\tag{91}\]
then,
\[q = -nF\,\Delta E \;-\; nF\,\Delta E_{\mathrm{appl}} \;+\; q_{\mathrm{rev}}\tag{92}\]
\[q = -nF\!\left( \Delta E + \Delta E_{\mathrm{appl}} \right) + q_{\mathrm{rev}}\tag{93}\]
When |ΔE| = ΔEappl and q = qrev, as we would expect; the applied voltage then just matches ΔE and the cell operates reversibly. As ΔEappl exceeds |ΔE| then excess heat begins to be evolved.
This discussion assumed an isothermal system. For an actual industrial cell the electrolyte temperature will rise and reach some steady state (although it may fluctuate with environmental conditions). There will be heat loss to the surroundings, but also heating of the electrolyte. Depending on the metal being electrowon, excess heat may need to be deliberately withdrawn by heat exchangers. The temperature of the solution will depend on the heat generated, loss to surroundings and the heat capacity of the solution.
Electrical work is supplied to the cell in order to overcome the cell's natural thermodynamic tendency. Some of that supplied energy does work on the cell, some of it ends up as heat. Some of the supplied work energy results in an increase in chemical potential energy. This is the net effect of breaking bonds (e.g. H-H bonds and Zn-O bonds in [Zn(H2O)6]+2, and forming new ones (e.g. Zn-Zn metal-metal bonds and H-O bonds in H3O+) and, finally, changes in electrostatic interactions in the solution due to changes in composition ([H+] increases; [Zn+2] decreases). Electrostatic interactions involve charged ions (Zn+2, H3O+, SO42-) and dipoles (such as partial charge separation in H-O-H, the oxygen being more electronegative and developing a negative charge; the hydrogens having a positive charge).
Rates of Electron Transfer and the Effects of an Applied Voltage
The rate of metal plating is directly proportional to the current through the cell, since,
\[I = \frac{C}{\text{s}}\;\propto\;\frac{\text{mol}\, e^-}{\text{s}}\;\propto\;\frac{\text{mol}\,\ce{Cu^{2+}}}{\text{s}} \text{reacted, etc.}\tag{94}\]
In electrowinning we set the current and allow the voltage to adjust accordingly (as governed by V = IR). Thus the higher the current, the higher the applied voltage must be. When the applied voltage precisely matches the thermodynamic cell voltage, ΔEappl = -ΔE, the cell operates reversibly and the reaction is infinitely slow. When ΔEappl > -ΔE the reaction proceeds at a finite rate, and the greater the difference the greater the rate.
Electrode polarity
For a spontaneous reaction electrons flow from (-) to (+); repelled from the negative electrode and attracted to the positive one. Hence the cathode is positively polarized and the anode is negative. This accords with the fact that the thermodynamic cell voltage is positive,
\[\Delta E = E_{\text{cathode}} - E_{\text{anode}} > 0 \tag{95}\]
In an electrolysis the applied voltage opposes the thermodynamic voltage (ΔE < 0) and is greater than -ΔE. Thus power supply (+) goes to the cell (+) and likewise the (-) of the power supply goes to (-) of the cell. This forces the cell to run in the opposite direction, so that now the cathode is negatively polarized and the anode is positive. In other words, the electrodes retain the same polarity in either the galvanic or the electrolytic cases, but the flow of electrons is opposite, as is the direction of the chemical reaction.
Media Attributions
- Ch7_F2_4_Electrochemical_Cells © Bé Wassink and Amir M. Dehkoda adapted by Jeno Hwang is licensed under a CC BY-NC (Attribution NonCommercial) license
- Ch7_F3_Zinc_HER_Slow © Bé Wassink and Amir M. Dehkoda is licensed under a CC BY-NC (Attribution NonCommercial) license
- Ch7_F4_Galvanic_Short-Circuit_Thermodynamics © Bé Wassink and Amir M. Dehkoda is licensed under a CC BY-NC (Attribution NonCommercial) license
- Ch7_F5_Alternatice_Thermodynamic_Effects © Bé Wassink and Amir M. Dehkoda is licensed under a CC BY-NC (Attribution NonCommercial) license
- Ch7_F6_Electrolytic_Cell_qrev © Bé Wassink and Amir M. Dehkoda is licensed under a CC BY-NC (Attribution NonCommercial) license
1. One compartment of an electrochemical cell, where either reduction or oxidation occurs. 2. A half reaction.
For electrochemical systems standard conditions are a combination of specified pressure, temperature and composition. Traditionally, 1 atm pressure for reactants and products and unit activities (molal scale) of the ions/compounds involved. Pure solids and liquids have unit activity by definition. Usually standard reduction potentials are tabulated for 25°C. Conversions to other temperature can be made. Some time ago, the pressure standard was changed to 1 bar = 100 kPa = 100,000 N/m2 = 0.98692 atm. Some care is needed to determine what conditions were employed when using a table of standard potentials.
