17 Voltammetric Methods
Voltammetric methods measure and plot current versus applied voltage. Voltammetric methods vary in how the voltage is applied in time.
Electrode Configuration
Whereas potentiometry frequently uses two-electrode cells (e.g. indicator and reference electrodes), voltammetry generally uses three-electrode cells with a reference electrode, working electrode, and an auxiliary (a.k.a. counter) electrode. The manner in which these electrodes are connected allows current to flow between the working electrode (where the redox reaction of interest occurs) and the counter electrode while the potential of the working electrode is measured against the reference electrode.
Common reference electrodes include silver/silver chloride and calomel. Common working electrodes include platinum, gold, carbon, and mercury. Other practical considerations for voltammetry include degassing the solution to minimize potential interference from dissolved oxygen, and the decision to stir or not stir the solution due to the impact of stirring on mass transport.
Voltammetric Methods
Linear Sweep Voltammetry (LSV). In LSV, the potential applied to the working electrode is linearly increased as a function of time. The current response is small (charging current only) until the onset potential for a Faradaic process is reached. Beyond this potential, the measured current increases to a peak and reaches a limiting current. The potential at which the current is half its peak value is the half-wave potential and is a function of the standard potential for the analyte-related redox reaction that occurs. The peak current is proportional to the concentration of analyte in a well-designed LSV method. The Randles-Sevcik equation relates the peak current (Eqn. 17.1) to physical constants (R, F), the temperature (T), the number of electrons transferred in the redox reaction (n), the electrode surface area (A), the analyte diffusion coefficient (D) and bulk concentration (c0), and the voltammetric scan rate (ν).
(Eqn. 17.1) [latex]I_{peak} = 0.4463(\frac{F^3}{RT})^{0.5}n^{1.5}AD^{0.5}ν^{0.5}c_0[/latex]
Although simple and the foundation of more advanced approaches, detection limits for LSV are modest (10–5–10–6 M) when compared to other voltammetric methods.
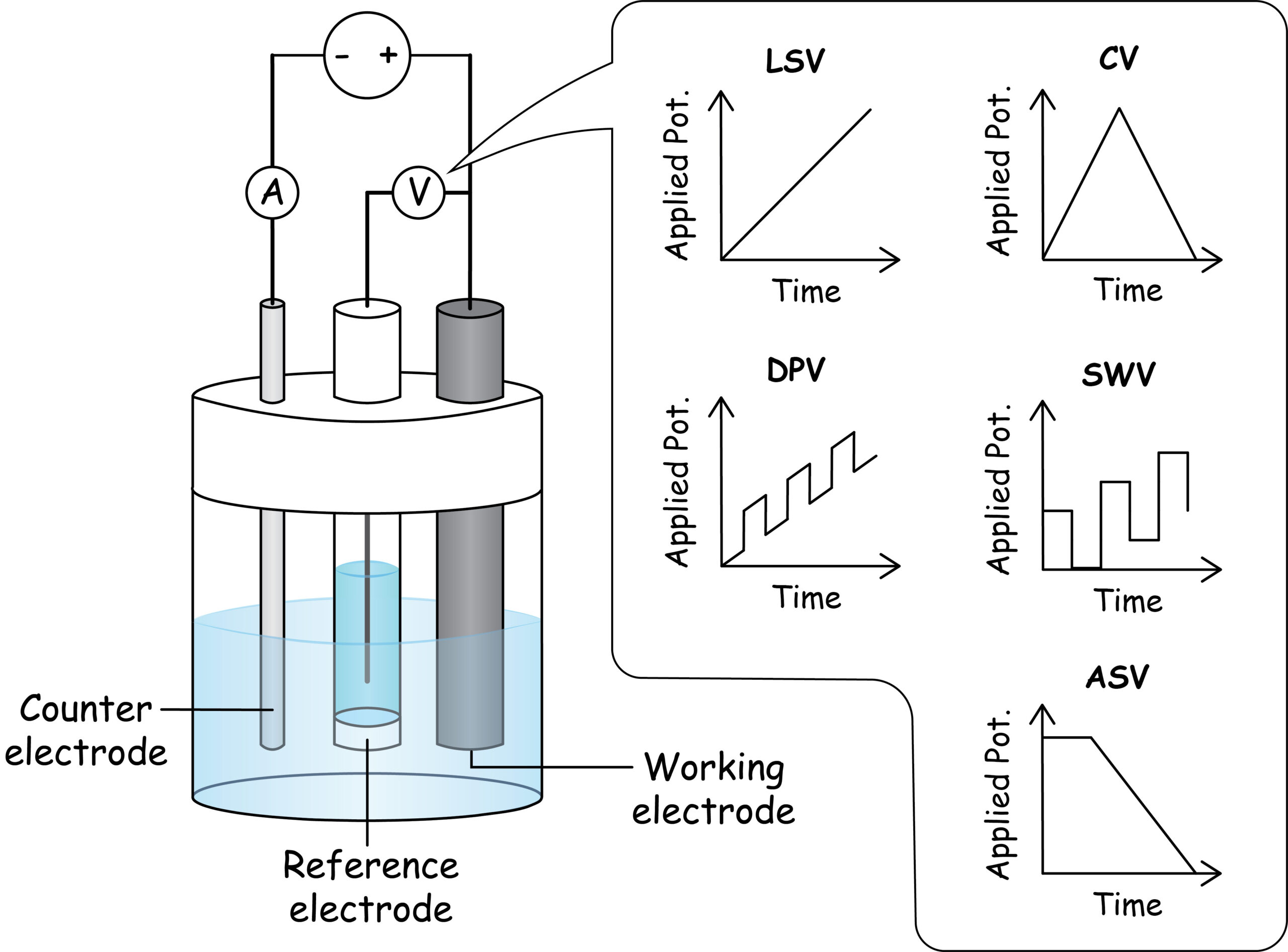
Cyclic Voltammetry (CV). In CV, a linear increase in potential over time is followed by a mirror-image linear decrease back to the starting potential. For reversible redox reactions, this method yields a “duck-shaped” plot of current versus voltage that partially resembles an LSV plot and its reflection. CV is not widely used for quantitative analysis, but is widely used for mechanistic and kinetic studies of redox reactions.
Differential Pulse Voltammetry (DPV). In DPV, short pulses of ‘extra’ voltage (e.g. 1–100 mV for 10–100 ms) are superimposed on a linear increase in applied potential with time. The current is measured immediately prior to the start of each pulse and just prior to the end of each pulse; the difference in current between these two points is plotted versus the applied linear sweep voltage. This approach largely isolates the Faradaic current from the charging current and yields plots with well-defined peaks. Square wave voltammetry (SWV) is conceptually similar to DPV and features a square wave of potential pulses superimposed on a staircase potential sweep. Detection limits of 10–7–10–8 M are common for pulsed voltammetry methods.
Anodic Stripping Voltammetry (ASV). For ASV, a negative potential is applied to the working electrode for an extended period (e.g. minutes to hours) and is followed by a potential sweep in the positive direction. These two stages are called deposition and stripping, respectively. Metal analytes are reduced onto the electrode during the deposition stage, then re-oxidized during the stripping stage. The potential sweep during stripping may be linear or pulsed (analogous to DPV or SWV). Because of the pre-concentration of analyte during deposition, ASV offers detection limits of 10–11 to 10–13 M for some metals.
Connections
- Scanning potential in voltammetry is analogous to scanning wavelength in optical spectroscopy (Ch. 7, Ch. 11).
- Analogous to UV-visible spectrophotometry (Ch. 7), a voltammetric scan of a blank sample is useful.
- The Randles-Sevcik equation has conceptual similarities to the Beer-Lambert law (Ch. 6): the electrode surface area and scan rate are instrument terms that are similar to the path length term in the Beer-Lambert law; and the analyte diffusion coefficient and number of electrons transferred are analyte properties that are similar to the molar absorption coefficient. Both equations predict a linear trend with analyte concentration.
Post-Reading Questions
- What pairs of electrodes are most relevant to measurements of (a) current and (b) potential in a voltammetric experiment?
- What are common working electrode materials?
- What are common types of reference electrodes?
- What method would have a lower detection limit: linear sweep voltammetry, ASV with a linear sweep stripping, or ASV with differential pulse stripping?
Topic Learning Objectives
The chapter is a primer for the following learning objectives, which will be covered in lecture and/or with additional assigned reading:
- Draw and label a diagram of a three-electrode cell.
- Draw and label diagrams of current versus potential and potential versus time for different voltammetric methods.
- Relate measured current to analyte concentration and measured potential to analyte identity.
- Explain how DPV and SWV overcome background from charging current.
- Discuss the origin of current peaks for DPV, SWV, and ASV.
- Discuss the origin of the different detection limits for different voltammetric methods.