Unit 6.2: Antimicrobial Drugs
Outline
Fundamentals of Antimicrobial Chemotherapy
Mechanisms of Antibacterial Drugs
- Inhibitors of Cell Wall Biosynthesis
- Inhibitors of Protein Biosynthesis
- Inhibitors of Membrane Function
- Inhibitors of Nucleic Acid Synthesis
- Inhibitors of Metabolic Pathways
Mechanisms of Other Antimicrobial Drugs
- Drug Modification or Inactivation
- Prevention of Cellular Uptake or Efflux
- Target Modification
- Other Modes of Resistance
Learning Objectives
After reading the following, you should be able to:
- Explain how antimicrobial drugs differ in use from antiseptics and disinfectants.
- Explain broad and narrow spectrum antibiotics and give an example of each.
- Describe the mode of action and give one example for each of the five major modes of action for antibiotics (Know penicillin, polymyxin, Rifamycin, sulfonamides and tetracycline).
- Explain the difficulty in development of drugs to target fungal, protozoan and helminthic infections.
- Describe the mechanism of action and give one example of an anti-fungal (miconazole), anti-protozoan (atovaquone) and anti-helminth (ivermectin) medication.
- Explain the difficulty in finding anti-viral drugs, and briefly explain the types of drugs that are in current use.
- Describe the Kirby-Bauer Technique.
- Describe how antibiotic resistance is a result of antibiotic usage.
- Describe four mechanisms through which microbes can resist drugs.
- Briefly describe phage therapy and its applications in treating diseases.
Ancient Chemotherapy: Most people associate the term chemotherapy with treatments for cancer. However, chemotherapy is actually a broader term that refers to any use of chemicals or drugs to treat disease. Chemotherapy may involve drugs that target cancerous cells or tissues, or it may involve antimicrobial drugs that target infectious microorganisms. Antimicrobial drugs typically work by destroying or interfering with microbial structures and enzymes, either killing microbial cells or inhibiting of their growth. But before we examine how these drugs work, we will briefly explore the history of humans’ use of antimicrobials for the purpose of chemotherapy
Although the discovery of antimicrobials and their subsequent widespread use is commonly associated with modern medicine, there is evidence that humans have been exposed to antimicrobial compounds for millennia. Chemical analyses of the skeletal remains of people from Nubia (now found in present-day Sudan) dating from between 350 and 550 AD have shown residue of the antimicrobial agent tetracycline in high enough quantities to suggest the purposeful fermentation of tetracycline-producing Streptomyces during the beer-making process. The resulting beer, which was thick and gruel-like, was used to treat a variety of ailments in both adults and children, including gum disease and wounds. The antimicrobial properties of certain plants may also have been recognized by various cultures around the world, including Indian and Chinese herbalists (Figure 6.16) who have long used plants for a wide variety of medical purposes. Healers of many cultures understood the antimicrobial properties of fungi and their use of moldy bread or other mold-containing products to treat wounds has been well documented for centuries. Today, while about 80% of the world’s population still relies on plant-derived medicines, scientists are now discovering the active compounds conferring the medicinal benefits contained in many of these traditionally used plants.

For a reminder of some of the scientists involved in the early days of modern chemotherapy, review unit 1.
Fundamentals of Antimicrobial Chemotherapy: An important distinction to remember is that where antiseptics and disinfectants are used to lower microbial counts to prevent infection, antimicrobial drugs are used for treating patients with infections. Using antimicrobials as preventatives is one of main causes leading to the prevalence of antibiotic resistant bacteria.
Several factors are important in choosing the most appropriate antimicrobial drug therapy, including bacteriostatic versus bactericidal mechanisms and spectrum of activity. (Dosage and route of administration, the potential for side effects, and the potential interactions between drugs are also important considerations for applying chemotherapy, but will not be discussed in this course.) The following discussion will focus primarily on antibacterial drugs, but the concepts translate to other antimicrobial classes.
Antibacterial drugs can be either bacteriostatic or bactericidal in their interactions with target bacteria. Bacteriostatic drugs cause a reversible inhibition of growth, with bacterial growth restarting after elimination of the drug. By contrast, bactericidal drugs kill their target bacteria. The decision of whether to use a bacteriostatic or bactericidal drugs depends on the type of infection and the immune status of the patient. In a patient with strong immune defenses, bacteriostatic and bactericidal drugs can be effective in achieving clinical cure. However, when a patient is immunocompromised, a bactericidal drug is essential for the successful treatment of infections. Regardless of the immune status of the patient, life-threatening infections such as acute endocarditis require the use of a bactericidal drug.
The spectrum of activity of an antibacterial drug relates to diversity of targeted bacteria. A narrow-spectrum antimicrobial targets only specific subsets of bacterial pathogens. For example, some narrow-spectrum drugs only target gram-positive bacteria, whereas others target only gram-negative bacteria. If the pathogen causing an infection has been identified, it is best to use a narrow-spectrum antimicrobial and minimize collateral damage to the normal microbiota. A broad-spectrum antimicrobial targets a wide variety of bacterial pathogens, including both gram-positive and gram-negative species, and is frequently used as empiric therapy to cover a wide range of potential pathogens while waiting on the laboratory identification of the infecting pathogen. Broad-spectrum antimicrobials are also used for polymicrobic infections (mixed infection with multiple bacterial species), or as prophylactic prevention of infections with surgery/invasive procedures. Finally, broad-spectrum antimicrobials may be selected to treat an infection when a narrow-spectrum drug fails because of development of drug resistance by the target pathogen.
The risk associated with using broad-spectrum antimicrobials is that they will also target a broad spectrum of the normal microbiota, increasing the risk of a superinfection, a secondary infection in a patient having a preexisting infection. A superinfection develops when the antibacterial intended for the preexisting infection kills the protective microbiota, allowing another pathogen resistant to the antibacterial to proliferate and cause a secondary infection (Figure 6.17). Common examples of superinfections that develop as a result of antimicrobial usage include yeast infections (candidiasis) and pseudomembranous colitis caused by Clostridioides difficile, which can be fatal.
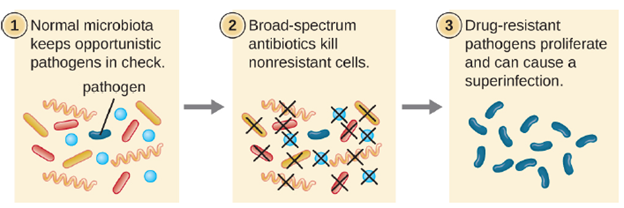
Mechanisms of Antibacterial Drugs: An important quality for an antimicrobial drug is selective toxicity, meaning that it selectively kills or inhibits the growth of microbial targets while causing minimal or no harm to the host. Most antimicrobial drugs currently in clinical use are antibacterial because the prokaryotic cell provides a greater variety of unique targets for selective toxicity, in comparison to fungi, parasites, and viruses. Each class of antibacterial drugs has a unique mode of action (the way in which a drug affects microbes at the cellular level), and these are summarized in Figure 6.18 and Table 6.1.
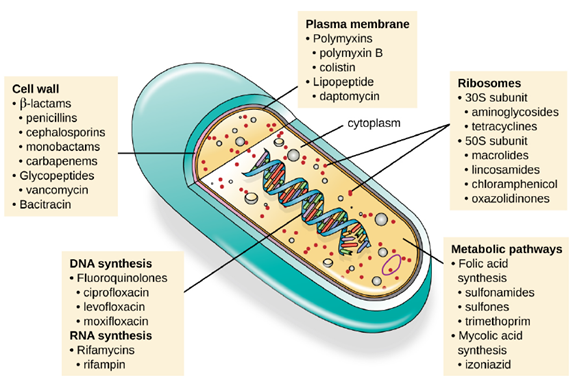
| Mode of Action | Target | Drug Class |
|---|---|---|
| Inhibit cell wall biosynthesis | Penicillin-binding proteins | beta-lactams: penicillins, cephalosporins, monobactams, carbapenems |
| Peptidoglycan subunits | Glycopeptides | |
| Peptidoglycan subunit transport | Bacitracin | |
| Inhibit biosynthesis of proteins | 30S ribosomal subunit | Aminoglycosides, tetracyclines |
| 50S ribosomal subunit | Macrolides, lincosamides, chloramphenicol, oxazolidinones | |
| Disrupt membranes | Lipopolysaccharide, inner and outer membranes | Polymyxin B, colistin, daptomycin |
| Inhibit nucleic acid synthesis | RNA | Rifamycin |
| DNA | Fluoroquinolones | |
| Antimetabolites | Folic acid synthesis enzyme | Sulfonamides, trimethoprim |
| Mycolic acid synthesis enzyme | Isonicotinic acid hydrazide | |
| Mycobacterial adenosine triphosphate (ATP) synthase inhibitor | Mycobacterial ATP synthase | Diarylquinoline |
1: Inhibitors of Cell Wall Biosynthesis: Several different classes of antibacterials block steps in the biosynthesis of peptidoglycan, making cells more susceptible to osmotic lysis. Therefore, antibacterials that target cell wall biosynthesis are bactericidal in their action. Because human cells do not make peptidoglycan, this mode of action is an excellent example of selective toxicity.
Penicillin, the first antibiotic discovered, is one of several antibacterials within a class called β-lactams. This group of compounds includes the penicillins, cephalosporins, monobactams, and carbapenems, and is characterized by the presence of a β-lactam ring found within the central structure of the drug molecule. The β-lactam antibacterials block the crosslinking of peptide chains during the biosynthesis of new peptidoglycan in the bacterial cell wall. Penicillin G and penicillin V are natural antibiotics from fungi and are primarily active against gram-positive bacterial pathogens, and a few gram-negative bacterial pathogens such as Pasteurella multocida.
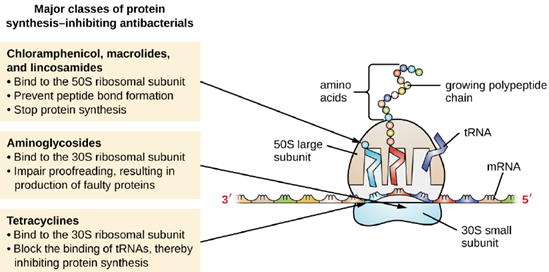
- Inhibitors of Protein Biosynthesis: The cytoplasmic ribosomes found in animal cells (80S) are structurally distinct from those found in bacterial cells (70S), making protein biosynthesis a good selective target for antibacterial drugs. Several types of protein biosynthesis inhibitors are discussed in this section and are summarized in Figure 6.19.
Aminoglycosides are large, highly polar antibacterial drugs that bind to the 30S subunit of bacterial ribosomes, impairing the proofreading ability of the ribosomal complex. This impairment causes the production of proteins with incorrect amino acids and shortened proteins that insert into the cytoplasmic membrane. Disruption of the cytoplasmic membrane by the faulty proteins kills the bacterial cells. The aminoglycosides, which include drugs such as streptomycin, gentamicin, neomycin, and kanamycin, are potent broad-spectrum antibacterials. However, aminoglycosides have been shown to be nephrotoxic (damaging to kidney), neurotoxic (damaging to the nervous system), and ototoxic (damaging to the ear).
Another class of antibacterial compounds that bind to the 30S subunit is the tetracyclines. In contrast to aminoglycosides, these drugs are bacteriostatic and inhibit protein synthesis by blocking the association of tRNAs with the ribosome during translation. Naturally occurring tetracyclines produced by various strains of Streptomyces were first discovered in the 1940s, and several semisynthetic tetracyclines, including doxycycline and tigecycline have also been produced. Although the tetracyclines are broad spectrum in their coverage of bacterial pathogens, side effects that can limit their use include phototoxicity, permanent discoloration of developing teeth, and liver toxicity with high doses or in patients with kidney impairment.
- Inhibitors of Membrane Function: A small group of antibacterials target the bacterial membrane as their mode of action. The polymyxins are natural polypeptide antibiotics that were first discovered in 1947 as products of Bacillus polymyxa; only polymyxin B and polymyxin E (colistin) have been used clinically. They are lipophilic with detergent-like properties and interact with the lipopolysaccharide component of the outer membrane of gram-negative bacteria, ultimately disrupting both their outer and inner membranes and killing the bacterial cells. Unfortunately, the membrane-targeting mechanism is not a selective toxicity, and these drugs also target and damage the membrane of cells in the kidney and nervous system when administered systemically. Because of these serious side effects and their poor absorption from the digestive tract, polymyxin B is used in over-the-counter topical antibiotic ointments (e.g., Neosporin), and oral colistin was historically used only for bowel decontamination to prevent infections originating from bowel microbes in immunocompromised patients or for those undergoing certain abdominal surgeries. However, the emergence and spread of multidrug-resistant pathogens has led to increased use of intravenous colistin in hospitals, often as a drug of last resort to treat serious infections. The antibacterial daptomycin is a cyclic lipopeptide produced by Streptomyces
roseosporus that seems to work like the polymyxins, inserting in the bacterial cell membrane and disrupting it. However, in contrast to polymyxin B and colistin, which target only gram-negative bacteria, daptomycin specifically targets gram-positive bacteria. It is typically administered intravenously and seems to be well tolerated, showing reversible toxicity in skeletal muscles.
- Inhibitors of Nucleic Acid Synthesis: Some antibacterial drugs work by inhibiting nucleic acid synthesis and are generally broad spectrum. For example, metronidazole is a semisynthetic member of the nitroimidazole family that is also an antiprotozoan. It interferes with DNA replication in target cells. The drug rifampin is a semisynthetic member of the rifamycin family and functions by blocking RNA polymerase activity in bacteria. The RNA polymerase enzymes in bacteria are structurally different from those in eukaryotes, providing for selective toxicity against bacterial cells. It is used for the treatment of a variety of infections, but its primary use, often in a cocktail with other antibacterial drugs, is against mycobacteria that cause tuberculosis. Despite the selectivity of its mechanism, rifampin can induce liver enzymes to increase metabolism of other drugs being administered (antagonism), leading to hepatotoxicity (liver toxicity) and negatively influencing the bioavailability and therapeutic effect of the companion drugs.
- Inhibitors of Metabolic Pathways: Some synthetic drugs control bacterial infections by functioning as antimetabolites, competitive inhibitors for bacterial metabolic enzymes. The sulfonamides (sulfa drugs) are the oldest synthetic antibacterialagents and are structural analogues of para-aminobenzoic acid (PABA), an early intermediate in folic acid synthesis. By inhibiting the enzyme involved in the production of dihydrofolic acid, sulfonamides block bacterial biosynthesis of folic acid and, subsequently, pyrimidines and purines required for nucleic acid synthesis. This mechanism of action provides bacteriostatic inhibition of growth against a wide spectrum of gram-positive and gram-negative pathogens. Because humans obtain folic acid from food instead of synthesizing it intracellularly,
sulfonamides are selectively toxic for bacteria. However, allergic reactions to sulfa drugs are common. The sulfones are structurally similar to sulfonamides but are not commonly used today except for the treatment of leprosy.
Mechanisms of Other Antimicrobial Drugs: Because fungi, protozoa, and helminths are eukaryotic, their cells are very similar to human cells, making it more difficult to develop drugs with selective toxicity. Additionally, viruses replicate within human host cells, making it difficult to develop drugs that are selectively toxic to viruses or virus-infected cells. Despite these challenges, there are antimicrobial drugs that target fungi, protozoa, helminths, and viruses, and some even target more than one type of microbe.
- Antifungal Drugs: The most common mode of action for antifungal drugs is the disruption of the cell membrane. Antifungals take advantage of small differences between fungi and humans in the biochemical pathways that synthesize sterols. The sterols are important in maintaining proper membrane fluidity and, hence, proper function of the cell membrane. For most fungi, the predominant membrane sterol is ergosterol. Because human cell membranes use cholesterol, instead of ergosterol, antifungal drugs that target ergosterol synthesis are selectively toxic.
The imidazoles are synthetic fungicides that disrupt ergosterol biosynthesis; they are commonly used in medical applications and also in agriculture to keep seeds and harvested crops from molding. Examples include miconazole, ketoconazole, and clotrimazole, which are used to treat fungal skin infections such as ringworm, specifically tinea pedis (athlete’s foot), tinea cruris (jock itch), and tinea corporis. These infections are commonly caused by dermatophytes of the genera Trichophyton, Epidermophyton, and Microsporum. Miconazole is also used predominantly for the treatment of vaginal yeast infections caused by the fungus Candida, and ketoconazole is used for the treatment of tinea versicolor and dandruff, which both can be caused by the fungus Malassezia.
- Antiprotozoan Drugs: There are a few mechanisms by which antiprotozoan drugs target infectious protozoans. Some are antimetabolites, such as atovaquone, proguanil, and artemisinins. Atovaquone, in addition to being antifungal, blocks electron transport in protozoans and is used for the treatment of protozoan infections including malaria, babesiosis, and toxoplasmosis. Proguanil is another synthetic antimetabolite that is processed in parasitic cells into its active form, which inhibits protozoan folic acid synthesis. It is often used in combination with atovaquone, and the combination is marketed as Malarone for both malaria treatment and prevention.
Artemisinin, a plant-derived antifungal first discovered by Chinese scientists in the 1970s, is quite effective against malaria. Semisynthetic derivatives of artemisinin are more water-soluble than the natural version, which makes them more bioavailable. Although the exact mechanism of action is unclear, artemisinins appear to act as prodrugs that are metabolized by target cells to produce reactive oxygen species (ROS) that damage target cells. Due to the rise in resistance to antimalarial drugs, artemisinins are also commonly used in combination with other antimalarial compounds in artemisinin-based combination therapy (ACT). Several antimetabolites are used for the treatment of toxoplasmosis caused by the parasite Toxoplasma gondii. The synthetic sulfa drug sulfadiazine competitively inhibits an enzyme in folic acid production in parasites and can be used to treat malaria and toxoplasmosis.
3: Antihelminthic Drugs: Because helminths are multicellular eukaryotes like humans, developing drugs with selective toxicity against them is extremely challenging. Despite this, several effective classes have been developed, including the avermectin family of drugs. The avermectins are members of the macrolide family that were first discovered from a Japanese soil isolate, Streptomyces avermectinius. A more potent semisynthetic derivative of avermectin is ivermectin, which binds to glutamate-gated chloride channels specific to invertebrates including helminths, blocking neuronal transmission and causing starvation, paralysis, and death of the worms. Ivermectin is used to treat roundworm diseases, including onchocerciasis (also called river blindness, caused by the worm Onchocerca volvulus) and strongyloidiasis (caused by the worm Strongyloides stercoralis or S. fuelleborni). Ivermectin also can also treat parasitic insects like mites, lice, and bed bugs, and is nontoxic to humans.
4: Antiviral Drugs: Unlike the complex structure of fungi, protozoa, and helminths, viral structure is simple, consisting of nucleic acid, a protein coat, viral enzymes, and, sometimes, a lipid envelope. Furthermore, viruses are obligate intracellular pathogens that use the host’s cellular machinery to replicate. These characteristics make it difficult to develop drugs with selective toxicity against viruses.
Neuraminidase inhibitors, including olsetamivir (Tamiflu), zanamivir (Relenza), and peramivir (Rapivab), specifically target influenza viruses by blocking the activity of influenza virus neuraminidase, preventing the release of the virus from infected cells. These three antivirals can decrease flu symptoms and shorten the duration of illness, but they differ in their modes of administration: olsetamivir is administered orally, zanamivir is inhaled, and peramivir is administered intravenously. Resistance to these neuraminidase inhibitors still seems to be minimal.
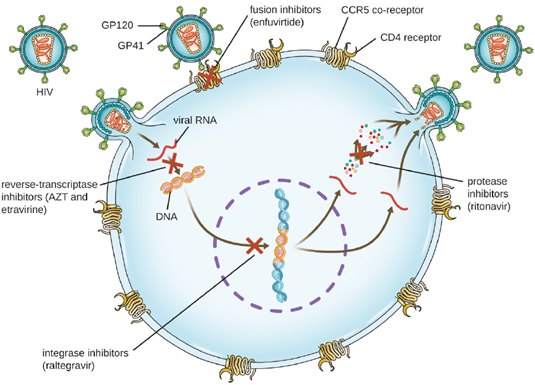
The reverse transcriptase inhibitors block the early step of converting viral RNA genome into DNA, and can include competitive nucleoside analog inhibitors (e.g., azidothymidine/zidovudine, or AZT) and non-nucleoside noncompetitive inhibitors (e.g., etravirine) that bind reverse transcriptase and cause an inactivating conformational change. Drugs called protease inhibitors (e.g., ritonavir) block the processing of viral proteins and prevent viral maturation. Protease inhibitors are also being developed for the treatment of other viral types. For example, simeprevir (Olysio) has been approved for the treatment of hepatitis C and is administered with ribavirin and interferon in combination therapy. The integrase inhibitors (e.g., raltegravir), block the activity of the HIV integrase responsible for the recombination of a DNA copy of the viral genome into the host cell chromosome. Additional drug classes for HIV treatment include the CCR5 antagonists and the fusion inhibitors (e.g., enfuviritide), which prevent the binding of HIV to the host cell coreceptor (chemokine receptor type 5 [CCR5]) and the merging of the viral envelope with the host cell membrane, respectively. Often, multiple antiviral medications are administered together as is the case with HIV treatments (Figure 6.20).
Another common treatment for chronic viral infections in the administration of interferons. Interferons of chemical mediators that are made as part of the body’s nature immune response in reaction to viral infections. These molecules can be synthesized artificially and given to patients to help treat viral infections. Interferons will be discussed in further detail in unit 8.
Antimicrobial Drug Resistance: Antimicrobial resistance is not a new phenomenon. In nature, microbes are constantly evolving in order to overcome the antimicrobial compounds produced by other microorganisms. Human development of antimicrobial drugs and their widespread clinical use has simply provided another selective pressure that promotes further evolution. Several important factors can accelerate the evolution of drug resistance. These include the overuse and misuse of antimicrobials, inappropriate use of antimicrobials, sub-therapeutic dosing, and patient noncompliance with the recommended course of treatment.
Exposure of a pathogen to an antimicrobial compound can select for chromosomal mutations conferring resistance, which can be transferred vertically to subsequent microbial generations and eventually become predominant in a microbial population that is repeatedly exposed to the antimicrobial. Alternatively, many genes responsible for drug resistance are found on plasmids or in transposons that can be transferred easily between microbes through horizontal gene transfer. Transposons also have the ability to move resistance genes between plasmids and chromosomes to further promote the spread of resistance. There are several common mechanisms for drug resistance, which are summarized in Figure 6.21. These mechanisms include enzymatic modification of the drug, modification of the antimicrobial target, and prevention of drug penetration or accumulation.
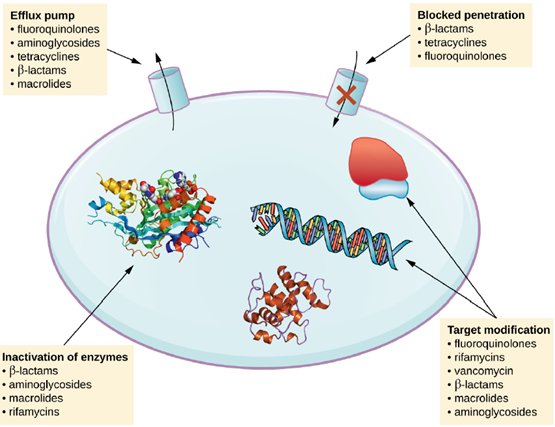
- Drug Modification or Inactivation: Resistance genes may code for enzymes that chemically modify an antimicrobial, thereby inactivating it, or destroy an antimicrobial through hydrolysis. Resistance to many types of antimicrobials occurs through this mechanism. For example, aminoglycoside resistance can occur through enzymatic transfer of chemical groups to the drug molecule, impairing the binding of the drug to its bacterial target. For β-lactams, bacterial resistance can involve the enzymatic hydrolysis of the β-lactam bond within the β-lactam ring of the drug molecule. Once the β-lactam bond is broken, the drug loses its antibacterial activity. This mechanism of resistance is mediated by β-lactamases, such as penicillinase, which are the most common mechanism of β-lactam resistance.
2/3. Prevention of Cellular Uptake or Efflux: Microbes may develop resistance mechanisms that involve inhibiting the accumulation of an antimicrobial drug, which then prevents the drug from reaching its cellular target. This strategy is common among gram-negative pathogens and can involve changes in outer membrane lipid composition, porin channel selectivity, and/or porin channel concentrations. Additionally, many gram-positive and gram-negative pathogenic bacteria produce efflux pumps that actively transport an antimicrobial drug out of the cell and prevent the accumulation of drug to a level that would be antibacterial. For example, resistance to β-lactams, tetracyclines, and fluoroquinolones commonly occurs through active efflux out of the cell, and it is rather common for a single efflux pump to have the ability to translocate multiple types of antimicrobials.
- Target Modification: Because antimicrobial drugs have very specific targets, structural changes to those targets can prevent drug binding, rendering the drug ineffective. Through spontaneous mutations in the genes encoding antibacterial drug targets, bacteria have an evolutionary advantage that allows them to develop resistance to drugs. This mechanism of resistance development is quite common. Genetic changes impacting the active site of penicillin-binding proteins (PBPs) can inhibit the binding of β-lactam drugs and provide resistance to multiple drugs within this class. This mechanism is very common among strains of Streptococcus pneumoniae, which alter their own PBPs through genetic mechanisms.
In contrast, strains of Staphylococcus aureus develop resistance to methicillin (MRSA) through the acquisition of a new low-affinity PBP, rather than structurally alter their existing PBPs. Not only does this new low-affinity PBP provide resistance to methicillin but it provides resistance to virtually all β-lactam drugs. Other examples of this resistance strategy include alterations in:
- ribosome subunits, providing resistance to macrolides, tetracyclines, and aminoglycosides;
- lipopolysaccharide (LPS) structure, providing resistance to polymyxins;
- RNA polymerase, providing resistance to rifampin;
- metabolic enzymes, providing resistance to sulfa drugs, sulfones, and trimethoprim
- Other modes of resistance: Other methods include overproduction of the target molecule, or by-passes for blocked pathways. A recently discovered mechanism of resistance called target mimicry involves the production of proteins that bind and sequester drugs, preventing the drugs from binding to their target. For example, Mycobacterium tuberculosis produces a protein with regular pentapeptide repeats that appears to mimic the structure of DNA. This protein binds fluoroquinolones, sequestering them and keeping them from binding to DNA, providing M. tuberculosis resistance to fluoroquinolones. Proteins that mimic the A-site of the bacterial ribosome have been found to contribute to aminoglycoside resistance as well.
Measuring Antimicrobial Effectiveness: Testing the effectiveness of antimicrobial drugs against specific organisms is important in identifying their spectrum of activity and the therapeutic dosage. The Kirby-Bauer disk diffusion test has long been used as a starting point for determining the susceptibility of specific microbes to various antimicrobial drugs. The Kirby-Bauer assay starts with a Mueller-Hinton agar plate on which a confluent lawn is inoculated with a patient’s isolated bacterial pathogen. Filter paper disks impregnated with known amounts of antibacterial drugs to be tested are then placed on the agar plate. As the bacterial inoculum grows, antibiotic diffuses from the circular disk into the agar and interacts with the growing bacteria. Antibacterial activity is observed as a clear circular zone of inhibition around the drug-impregnated disk, similar to the disk-diffusion assay depicted in Figure 6.15. The diameter of the zone of inhibition, measured in millimeters and compared to a standardized chart, determines the susceptibility or resistance of the bacterial pathogen to the drug.
There are multiple factors that determine the size of a zone of inhibition in this assay, including drug solubility, rate of drug diffusion through agar, the thickness of the agar medium, and the drug concentration impregnated into the disk. Due to a lack of standardization of these factors, interpretation of the Kirby-Bauer disk diffusion assay provides only limited information on susceptibility and resistance to the drugs tested. The assay cannot distinguish between bacteriostatic and bactericidal activities, and differences in zone sizes cannot be used to compare drug potencies or efficacies. Comparison of zone sizes to a standardized chart will only provide information on the antibacterials to which a bacterial pathogen is susceptible or resistant.
Phage therapy: With the increase in the prevalence of antibiotic resistant bacteria, it is becoming increasingly difficult to treat certain types of infections. Currently there are multiple pathogenic bacterial species that do not respond to any of the common antibacterial medications and the number of these will only continue to grow. As antibacterial medications are no longer always an option for treating certain diseases, other methods of treatment need to be used. One such method, gaining in popularity, is phage therapy.
Phage therapy involves the collection and growth of bacteriophages that target the pathogen of interest. These phages are traditionally taken orally or applied topically to affected areas. Intravenous application trials are currently underway in the United States and if successful, would greatly expand the number of infections that could be targeted using phage therapy. As discussed in unit 3, the phages infect the bacterial pathogens and result in their lysis, thus eliminating the pathogen and aiding in the patient’s recovery.
Since the bacteriophages cannot target animal cells, there is no concern about off-target toxicity in the patient. Also, since the administered phages are selected to target the pathogen and viruses have a specific host range, the phages tend to be less harmful to the patient’s microbiome, when compared to traditional antibiotics. Another advantage of phage therapies, is while bacteria may evolve resistance the phages, the phages themselves are also capable of evolution, allowing them to change along with their host and preventing resistance from occurring.
