Unit 8.2: Adaptive Immunity
Outline
Part 1: Overview of Specific Adaptive Immunity
Part 2: Major Histocompatibility Complexes and Antigen Presenting Cells
Part 3: T Lymphocytes and Cellular Immunity
- T Cell Production and Maturation
- Classes of T cells
- Activation and Differentiation of Helper T Cells
- Activation and Differentiation of Cytotoxic T Cells
Learning Objectives
After reading the following, you should be able to:
- Describe two differences between innate and specific immunity.
- Define: humoral vs. cell-mediated immunity, antibodies, antigen, epitopes, hapten
- Describe the activation of a helper T cell and the function of a helper T cell.
- Describe and give an example of cytokines important to the adaptive immune system.
- Describe the activation of a B cell against T-independent, and against T-dependent antigens and the function of a B cell.
- Describe the structure and function the five types of antibodies.
- Explain how a cytotoxic T cell is activated and the function of a cytotoxic T cell.
- Describe the two ways that a cytotoxic T cell can kill a body cell.
- Explain why a secondary response is much stronger than a primary response to a specific antigen
- Describe the function of vaccinations.
- Name and describe the different types of vaccines.
Part 1: Overview of Specific Adaptive Immunity
Adaptive immunity, often considered the third line of immune defense, is defined by two important characteristics: specificity and memory. Specificity refers to the adaptive immune system’s ability to target specific pathogens, and memory refers to its ability to quickly respond to pathogens to which it has previously been exposed. For example, when an individual recovers from chickenpox, the body develops a memory of the infection that will specifically protect it from the causative agent, the varicella-zoster virus, if it is exposed to the virus again later.
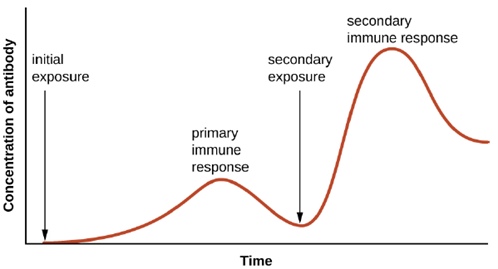
Specificity and memory are achieved by essentially programming certain cells involved in the immune response to respond rapidly to subsequent exposures of the pathogen. This programming occurs as a result of the first exposure to a pathogen or vaccine, which triggers a primary response. Subsequent exposures result in a secondary response that is faster and stronger as a result of the body’s memory of the first exposure (Figure 8.20). This secondary response, however, is specific to the pathogen in question. For example, exposure to one virus (e.g., varicella-zoster virus) will not provide protection against other viral diseases (e.g., measles, mumps, or polio).
Adaptive specific immunity involves the actions of two distinct cell types: B lymphocytes (B cells) and T lymphocytes (T cells). Although B cells and T cells arise from a common hematopoietic stem cell differentiation pathway (see Figure 8.10), their sites of maturation and their roles in adaptive immunity are very different.
B cells mature in the bone marrow and are responsible for the production of glycoproteins called antibodies, or immunoglobulins. Antibodies are involved in the body’s defense against pathogens and toxins in the extracellular environment. Mechanisms of adaptive specific immunity that involve B cells and antibody production are referred to as humoral immunity. The maturation of T cells occurs in the thymus. T cells function as the central orchestrator of both innate and adaptive immune responses. They are also responsible for destruction of cells infected with intracellular pathogens. The targeting and destruction of intracellular pathogens by T cells is called cell-mediated immunity, or cellular immunity.
Antigens: Activation of the adaptive immune defenses is triggered by pathogen-specific molecular structures called antigens. Antigens are similar to the pathogen-associated molecular patterns (PAMPs) discussed previously; however, whereas PAMPs are molecular structures found on numerous pathogens, antigens are unique to a specific pathogen. The antigens that stimulate adaptive immunity to chickenpox, for example, are unique to the varicella-zoster virus but significantly different from the antigens associated with other viral pathogens.
The term antigen was initially used to describe molecules that stimulate the production of antibodies; in fact, the term comes from a combination of the words antibody and generator, and a molecule that stimulates antibody production is said to be antigenic. However, the role of antigens is not limited to humoral immunity and the production of antibodies; antigens also play an essential role in stimulating cellular immunity, and for this reason antigens are sometimes more accurately referred to as immunogens. In this text, however, we will typically refer to them as antigens.
Pathogens possess a variety of structures that may contain antigens. For example, antigens from bacterial cells may be associated with their capsules, cell walls, fimbriae, flagella, or pili. Bacterial antigens may also be associated with extracellular toxins and enzymes that they secrete. Viruses possess a variety of antigens associated with their capsids, envelopes, and the spike structures they use for attachment to cells.
Antigens may belong to any number of molecular classes, including carbohydrates, lipids, nucleic acids, proteins, and combinations of these molecules. Antigens of different classes vary in their ability to stimulate adaptive immune defenses as well as in the type of response they stimulate (humoral or cellular). The structural complexity of an antigenic molecule is an important factor in its antigenic potential. In general, more complex molecules are more effective as antigens. For example, the three-dimensional complex structure of proteins make them the most effective and potent antigens, capable of stimulating both humoral and cellular immunity. In comparison, carbohydrates are less complex in structure and therefore less effective as antigens; they can only stimulate humoral immune defenses.
Lipids and nucleic acids are the least antigenic molecules, and in some cases may only become antigenic when combined with proteins or carbohydrates to form glycolipids, lipoproteins, or nucleoproteins.
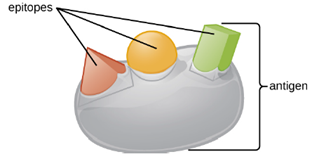
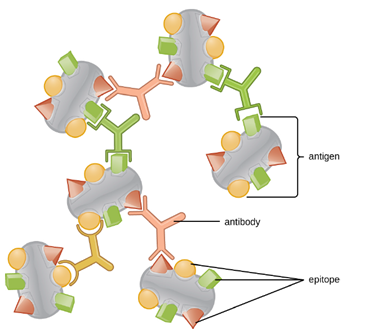
One reason the three-dimensional complexity of antigens is so important is that antibodies and T cells do not recognize and interact with an entire antigen but with smaller exposed regions on the surface of antigens called epitopes. A single antigen may possess several different epitopes (Figure 8.21), and different antibodies may bind to different epitopes on the same antigen (Figure 8.22). For example, the bacterial flagellum is a large, complex protein structure that can possess hundreds or even thousands of epitopes with unique three-dimensional structures. Moreover, flagella from different bacterial species (or even strains of the same species) contain unique epitopes that can only be bound by specific antibodies.
An antigen’s size is another important factor in its antigenic potential. Whereas large antigenic structures like flagella possess multiple epitopes, some molecules are too small to be antigenic by themselves. Such molecules, called haptens, are essentially free epitopes that are not part of the complex three-dimensional structure of a larger antigen. For a hapten to become antigenic, it must first attach to a larger carrier molecule (usually a protein) to produce a conjugate antigen. The hapten-specific antibodies produced in response to the conjugate antigen are then able to interact with unconjugated free hapten molecules. Haptens are not known to be associated with any specific pathogens, but they are responsible for some allergic responses. For example, the hapten urushiol, a molecule found in the oil of plants that cause poison ivy, causes an immune response that can result in a severe rash (called contact dermatitis). Similarly, the hapten penicillin can cause allergic reactions to drugs in the penicillin class.
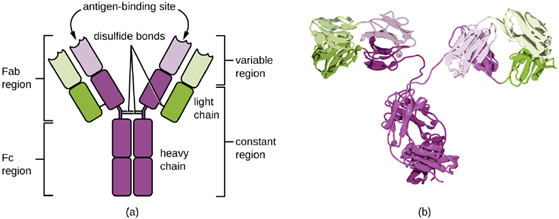
Antibodies: Antibodies (also called immunoglobulins) are glycoproteins that are present in both the blood and tissue fluids. The basic structure of an antibody monomer consists of four protein chains held together by disulfide bonds (Figure 8.23). A disulfide bond is a covalent bond between the sulfhydryl R groups found on two cysteine amino acids. The two largest chains are identical to each other and are called the heavy chains. The two smaller chains are also identical to each other and are called the light chains. Joined together, the heavy and light chains form a basic Y-shaped structure.
The two ‘arms’ of the Y-shaped antibody molecule are known as the Fab region, for “fragment of antigen binding.” The far end of the Fab region is the variable region, which serves as the site of antigen binding. The amino acid sequence in the variable region dictates the three-dimensional structure, and thus the specific three-dimensional epitope to which the Fab region is capable of binding. Although the epitope specificity of the Fab regions is identical for each arm of a single antibody molecule, this region displays a high degree of variability between antibodies with different epitope specificities. Binding to the Fab region is necessary for neutralization of pathogens, agglutination or aggregation of pathogens, and antibody-dependent cell-mediated cytotoxicity
The constant region of the antibody molecule includes the trunk of the Y and lower portion of each arm of the Y. The trunk of the Y is also called the Fc region, for “fragment of crystallization,” and is the site of complement factor binding and binding to phagocytic cells during antibody-mediated opsonization.
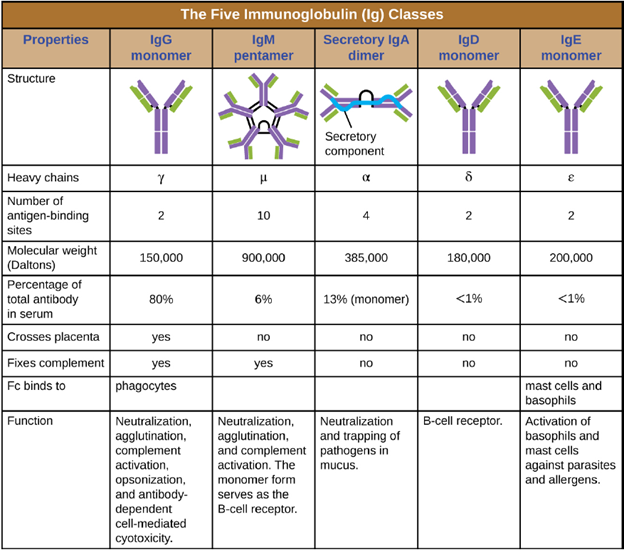
Antibody Classes: The constant region of an antibody molecule determines its class, or isotype. The five classes of antibodies are IgG, IgM, IgA, IgD, and IgE. Each class possesses unique heavy chains designated by Greek letters γ, μ, α, δ, and ε, respectively. Antibody classes also exhibit important differences in abundance in serum, arrangement, body sites of action, functional roles, and size (Figure 8.24).
IgG is a monomer that is by far the most abundant antibody in human blood, accounting for about 80% of total serum antibody. IgG penetrates efficiently into tissue spaces, and is the only antibody class with the ability to cross the placental barrier, providing passive immunity to the developing fetus during pregnancy. IgG is also the most versatile antibody class in terms of its role in the body’s defense against pathogens.
IgM is initially produced in a monomeric membrane-bound form that serves as an antigen-binding receptor on B cells. The secreted form of IgM assembles into a pentamer with five monomers of IgM bound together by a protein structure called the J chain. Although the location of the J chain relative to the Fc regions of the five monomers prevents IgM from performing some of the functions of IgG, the ten available Fab sites associated with a pentameric IgM make it an important antibody in the body’s arsenal of defenses. IgM is the first antibody produced and secreted by B cells during the primary and secondary immune responses, making pathogen-specific IgM a valuable diagnostic marker during active or recent infections
IgA accounts for about 13% of total serum antibody, and secretory IgA is the most common and abundant antibody class found in the mucus secretions that protect the mucous membranes. IgA can also be found in other secretions such as breast milk, tears, and saliva. Secretory IgA is assembled into a dimeric form with two monomers joined by a protein structure called the secretory component. One of the important functions of secretory IgA is to trap pathogens in mucus so that they can later be eliminated from the body.
Similar to IgM, IgD is a membrane-bound monomer found on the surface of B cells, where it serves as an antigenbinding receptor. However, IgD is not secreted by B cells, and only trace amounts are detected in serum. These trace amounts most likely come from the degradation of old B cells and the release of IgD molecules from their cytoplasmic membranes.
IgE is the least abundant antibody class in serum. Like IgG, it is secreted as a monomer, but its role in adaptive immunity is restricted to anti-parasitic defenses. The Fc region of IgE binds to basophils and mast cells. The Fab region of the bound IgE then interacts with specific antigen epitopes, causing the cells to release potent proinflammatory mediators. The inflammatory reaction resulting from the activation of mast cells and basophils aids in the defense against parasites, but this reaction is also central to allergic reactions.
Antigen-Antibody Interactions: Different classes of antibody play important roles in the body’s defense against pathogens. These functions include neutralization of pathogens, opsonization for phagocytosis, agglutination, complement activation, and antibody-dependent cell-mediated cytotoxicity. For most of these functions, antibodies also provide an important link between adaptive specific immunity and innate nonspecific immunity.
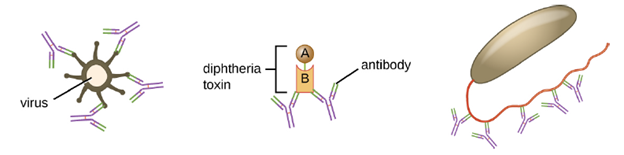
Neutralization involves the binding of certain antibodies (IgG, IgM, or IgA) to epitopes on the surface of pathogens or toxins, preventing their attachment to cells. For example, Secretory IgA can bind to specific pathogens and block initial attachment to intestinal mucosal cells. Similarly, specific antibodies can bind to certain toxins, blocking them from attaching to target cells and thus neutralizing their toxic effects. Viruses can be neutralized and prevented from infecting a cell by the same mechanism (Figure 8.25).
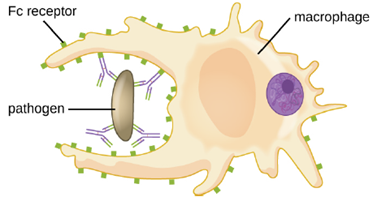
Opsonization is the coating of a pathogen with molecules, such as complement factors, C-reactive protein, and serum amyloid A, to assist in phagocyte binding to facilitate phagocytosis. IgG antibodies also serve as excellent opsonins, binding their Fab sites to specific epitopes on the surface of pathogens.
Phagocytic cells such as macrophages, dendritic cells, and neutrophils have receptors on their surfaces that recognize and bind to the Fc portion of the IgG molecules; thus, IgG helps such phagocytes attach to and engulf the pathogens they have bound (Figure 8.26).
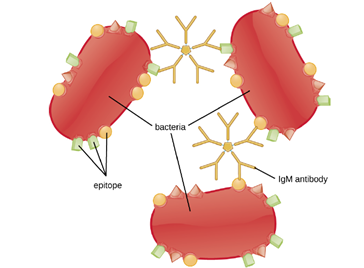
Agglutination or aggregation involves the cross-linking of pathogens by antibodies to create large aggregates (Figure 8.27). IgG has two Fab antigen-binding sites, which can bind to two separate pathogen cells, clumping them together. When multiple IgG antibodies are involved, large aggregates can develop; these aggregates are easier for the kidneys and spleen to filter from the blood and easier for phagocytes to ingest for destruction. The pentameric structure of IgM provides ten Fab binding sites per molecule, making it the most efficient antibody for agglutination.
Another important function of antibodies is activation of the complement cascade. As discussed in the innate immunity section, the complement system is an important component of the innate defenses, promoting the inflammatory response, recruiting phagocytes to site of infection, enhancing phagocytosis by opsonization, and killing gramnegative bacterial pathogens with the membrane attack complex (MAC). Complement activation can occur through three different pathways (see Figure 8.7), but the most efficient is the classical pathway, which requires the initial binding of IgG or IgM antibodies to the surface of a pathogen cell, allowing for recruitment and activation of the C1 complex.
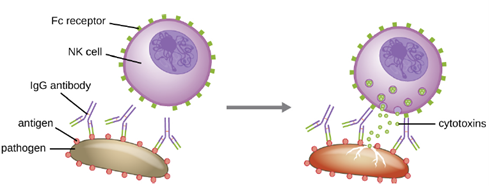
Yet another important function of antibodies is antibody-dependent cell-mediated cytotoxicity (ADCC), which enhances killing of pathogens that are too large to be phagocytized. This process is best characterized for natural killer cells (NK cells), as shown in Figure 8.28, but it can also involve macrophages and eosinophils. ADCC occurs when the Fab region of an IgG antibody binds to a large pathogen; Fc receptors on effector cells (e.g., NK cells) then bind to the Fc region of the antibody, bringing them into close proximity with the target pathogen. The effector cell then secretes powerful cytotoxins (e.g., perforin and granzymes) that kill the pathogen.
Part 2: Major Histocompatibility Complexes and Antigen- Presenting Cells
As discussed in Cellular Defenses, major histocompatibility complex (MHC) molecules are expressed on the surface of healthy cells, identifying them as normal and “self” to natural killer (NK) cells. MHC molecules also play an important role in the presentation of foreign antigens, which is a critical step in the activation of T cells and thus an important mechanism of the adaptive immune system.
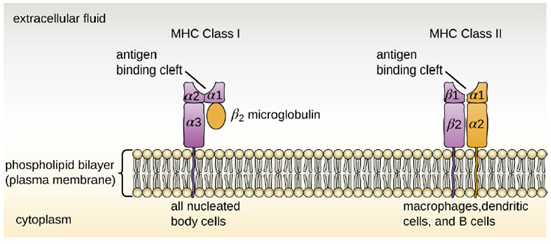
Major Histocompatibility Complex Molecules: The major histocompatibility complex (MHC) is a collection of genes coding for MHC molecules found on the surface of all nucleated cells of the body. Mature red blood cells, which lack a nucleus, are the only cells that do not express MHC molecules on their surface. There are two classes of MHC molecules involved in adaptive immunity, MHC I and MHC II (Figure 8.29). MHC I molecules are found on all nucleated cells; they present normal self-antigens as well as abnormal or nonself pathogens to the effector T cells involved in cellular immunity. In contrast, MHC II molecules are only found on macrophages, dendritic cells, and B cells; they present abnormal or nonself pathogen antigens for the initial activation of T cells.
Antigen-Presenting Cells (APCs): All nucleated cells in the body have mechanisms for processing and presenting antigens in association with MHC molecules. This signals the immune system, indicating whether the cell is normal and healthy or infected with an intracellular pathogen. However, only macrophages, dendritic cells, and B cells have the ability to present antigens specifically for the purpose of activating T cells; for this reason, these types of cells are sometimes referred to as antigen-presenting cells (APCs).
While all APCs play a similar role in adaptive immunity, there are some important differences to consider. Macrophages and dendritic cells are phagocytes that ingest and kill pathogens that penetrate the first-line barriers (i.e., skin and mucous membranes). B cells, on the other hand, do not function as phagocytes but play a primary role in the production and secretion of antibodies. In addition, whereas macrophages and dendritic cells recognize pathogens through nonspecific receptor interactions (e.g., PAMPs, toll-like receptors, and receptors for opsonizing complement or antibody), B cells interact with foreign pathogens or their free antigens using antigen-specific immunoglobulin as receptors (monomeric IgD and IgM). When the immunoglobulin receptors bind to an antigen, the B cell internalizes the antigen by endocytosis before processing and presenting the antigen to T cells.
1. Antigen Presentation with MHC II Molecules: MHC II molecules are only found on the surface of APCs. Macrophages and dendritic cells use similar mechanisms for processing and presentation of antigens and their epitopes in association with MHC II; B cells use somewhat different mechanisms that will be described further down. For now, we will focus on the steps of the process as they pertain to dendritic cells.
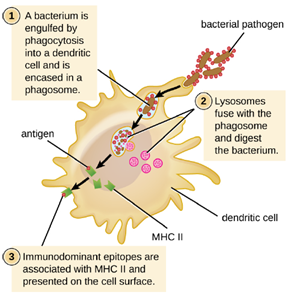
After a dendritic cell recognizes and attaches to a pathogen cell, the pathogen is internalized by phagocytosis and is initially contained within a phagosome. Lysosomes containing antimicrobial enzymes and chemicals fuse with the phagosome to create a phagolysosome, where degradation of the pathogen for antigen processing begins. Proteases (protein-degrading) are especially important in antigen processing because only protein antigen epitopes are presented to T cells by MHC II (Figure 8.30).
APCs do not present all possible epitopes to T cells; only a selection of the most antigenic or immunodominant epitopes are presented. The mechanism by which epitopes are selected for processing and presentation by an APC is complicated and not well understood; however, once the most antigenic, immunodominant epitopes have been processed, they associate within the antigen-binding cleft of MHC II molecules and are translocated to the cell surface of the dendritic cell for presentation to T cells.
2. Antigen Presentation with MHC I Molecules: MHC I molecules, found on all normal, healthy, nucleated cells, signal to the immune system that the cell is a normal “self” cell. In a healthy cell, proteins normally found in the cytoplasm are degraded by proteasomes (enzyme complexes responsible for degradation and processing of proteins) and processed into self-antigen epitopes; these self-antigen epitopes bind within the MHC I antigen-binding cleft and are then presented on the cell surface. Immune cells, such as NK cells, recognize these self-antigens and do not target the cell for destruction. However, if a cell becomes infected with an intracellular pathogen (e.g., a virus), protein antigens specific to the pathogen are processed in the proteasomes and bind with MHC I molecules for presentation on the cell surface. This presentation of pathogen-specific antigens with MHC I signals that the infected cell must be targeted for destruction along with the pathogen.
Part 3: T Lymphocytes and Cellular Immunity
As explained in above, the antibodies involved in humoral immunity often bind pathogens and toxins before they can attach to and invade host cells. Thus, humoral immunity is primarily concerned with fighting pathogens in extracellular spaces. However, pathogens that have already gained entry to host cells are largely protected from the humoral antibody-mediated defenses. Cellular immunity, on the other hand, targets and eliminates intracellular pathogens through the actions of T lymphocytes, or T cells (Figure 8.31). T cells also play a more central role in orchestrating the overall adaptive immune response (humoral as well as cellular) along with the cellular defenses of innate immunity.
T Cell Production and Maturation: T cells, like all other white blood cells involved in innate and adaptive immunity, are formed from multipotent hematopoietic stem cells (HSCs) in the bone marrow (see Figure 8.10). However, unlike the white blood cells of innate immunity, eventual T cells differentiate first into lymphoid stem cells that then become small, immature lymphocytes, sometimes called lymphoblasts. The first steps of differentiation occur in the red marrow of bones (Figure 8.32), after which immature T lymphocytes enter the bloodstream and travel to the thymus for the final steps of maturation (Figure 18.15). Once in the thymus, the immature T lymphocytes are referred to as thymocytes.
The maturation of thymocytes within the thymus can be divided into three critical steps of positive and negative selection, collectively referred to as thymic selection. The first step of thymic selection occurs in the cortex of the thymus and involves the development of a functional T-cell receptor (TCR) that is required for activation by APCs. Thymocytes with defective TCRs are removed by negative selection through the induction of apoptosis (programmed controlled cell death). The second step of thymic selection also occurs in the cortex and involves the positive selection of thymocytes that will interact appropriately with MHC molecules. Thymocytes that can interact appropriately with MHC molecules receive a positive stimulation that moves them further through the process of maturation, whereas thymocytes that do not interact appropriately are not stimulated and are eliminated by apoptosis. The third and final step of thymic selection occurs in both the cortex and medulla and involves negative selection to remove self-reacting thymocytes, those that react to self-antigens, by apoptosis. This final step is sometimes referred to as central tolerance because it prevents self-reacting T cells from reaching the bloodstream and potentially causing autoimmune disease, which occurs when the immune system attacks healthy “self” cells.
It has been estimated that the three steps of thymic selection eliminate 98% of thymocytes. The remaining 2% that exit the thymus migrate through the bloodstream and lymphatic system to sites of secondary lymphoid organs/tissues, such as the lymph nodes, spleen, and tonsils (Figure 18.15), where they await activation through the presentation of specific antigens by APCs. Until they are activated, they are known as mature naïve T cells.
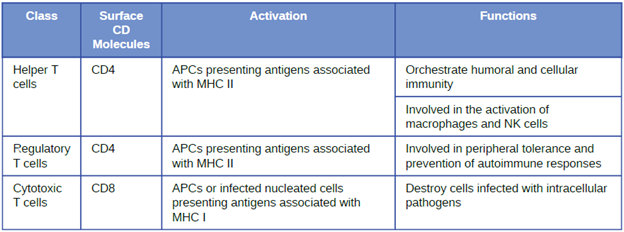
Classes of T Cells: T cells can be categorized into three distinct classes: helper T cells, regulatory T cells, and cytotoxic T cells. These classes are differentiated based on their expression of certain surface molecules, their mode of activation, and their functional roles in adaptive immunity (Table 8.6).
All T cells produce cluster of differentiation (CD) molecules, cell surface glycoproteins that can be used to identify and distinguish between the various types of white blood cells. Although T cells can produce a variety of CD molecules, CD4 and CD8 are the two most important used for differentiation of the classes. Helper T cells and regulatory T cells are characterized by the expression of CD4 on their surface, whereas cytotoxic T cells are characterized by the expression of CD8.
Classes of T cells can also be distinguished by the specific MHC molecules and APCs with which they interact for activation. Helper T cells and regulatory T cells can only be activated by APCs presenting antigens associated with MHC II. In contrast, cytotoxic T cells recognize antigens presented in association with MHC I, either by APCs or by nucleated cells infected with an intracellular pathogen.
The different classes of T cells also play different functional roles in the immune system. Helper T cells serve as the central orchestrators that help activate and direct functions of humoral and cellular immunity. In addition, helper T cells enhance the pathogen-killing functions of macrophages and NK cells of innate immunity. In contrast, the primary role of regulatory T cells is to prevent undesirable and potentially damaging immune responses. They play a role in peripheral tolerance, which is the inhibition of self-reactive T-cells that somehow survive thymic selection and exit the thymus. Finally, cytotoxic T cells are the primary effector cells for cellular immunity. They recognize and target cells that have been infected by intracellular pathogens, destroying infected cells along with the pathogens inside.
Activation and Differentiation of Helper T Cells: For both helper T cells and cytotoxic T cells, activation is a complex process that requires the interactions of multiple molecules and exposure to cytokines. The T-cell receptor (TCR) is involved in the first step of pathogen epitope recognition during the activation process. The TCR comes from the same receptor family as the antibodies IgD and IgM, the antigen receptors on the B cell membrane surface, and thus shares common structural elements. Similar to antibodies, the TCR has a variable region and a constant region, and the variable region provides the antigen-binding site. TCRs are epitope-specific, and it has been estimated that 25 million T cells with unique epitope-binding TCRs are required to protect an individual against a wide range of microbial pathogens.
Helper T cells can only be activated by APCs presenting processed foreign epitopes in association with MHC II. The first step in the activation process is TCR recognition of the specific foreign epitope presented within the MHC II antigen-binding cleft. The second step involves the interaction of CD4 on the helper T cell with a region of the MHC II molecule separate from the antigen-binding cleft. This second interaction anchors the MHC II-TCR complex and ensures that the helper T cell is recognizing both the foreign (“nonself”) epitope and “self” antigen of the APC; both recognitions are required for activation of the cell. In the third step, the APC and T cell secrete cytokines that activate the helper T cell. The activated helper T cell then proliferates, dividing by mitosis to produce clonal naïve helper T cells that differentiate into subtypes with different functions (Figure 8.33).
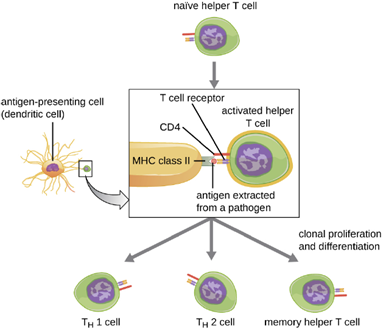
Activated helper T cells can differentiate into different subtypes. The differentiation process is directed by APC-secreted cytokines. Depending on which APC-secreted cytokines interact with an activated helper T cell, the cell may differentiate into a T helper 1 (TH1) cell, a T helper 2 (TH2) cell, or a memory helper T cell. The two types of helper T cells are relatively short-lived effector cells, meaning that they perform various functions of the immediate immune response. In contrast, memory helper T cells are relatively long lived; they are programmed to “remember” a specific antigen or epitope in order to mount a rapid, strong, secondary response to subsequent exposures.
TH1 cells secrete their own cytokines that are involved in stimulating and orchestrating other cells involved in adaptive and innate immunity. For example, they stimulate cytotoxic T cells, enhancing their killing of infected cells and promoting differentiation into memory cytotoxic T cells. TH1 cells also stimulate macrophages and neutrophils to become more effective in their killing of intracellular bacteria. They can also stimulate NK cells to become more effective at killing target cells.
TH2 cells play an important role in orchestrating the humoral immune response through their secretion of cytokines that activate B cells and direct B cell differentiation and antibody production. Various cytokines produced by TH2 cells orchestrate antibody class switching, which allows B cells to switch between the production of IgM, IgG, IgA, and IgE as needed to carry out specific antibody functions and to provide pathogen-specific humoral immune responses.
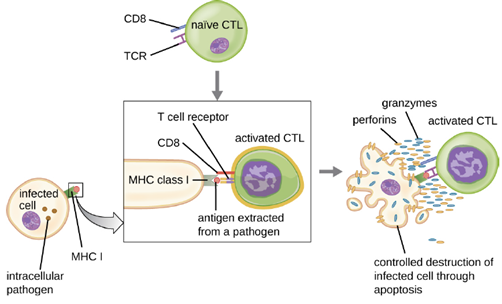
Activation and Differentiation of Cytotoxic T Cells: Cytotoxic T cells (also referred to as cytotoxic T lymphocytes, or CTLs) are activated by APCs in a three-step process similar to that of helper T cells. The key difference is that the activation of cytotoxic T cells involves recognition of an antigen presented with MHC I (as opposed to MHC II) and interaction of CD8 (as opposed to CD4) with the receptor complex. After the successful co-recognition of foreign epitope and self-antigen, the production of cytokines by the APC and the cytotoxic T cell activate clonal proliferation and differentiation. Activated cytotoxic T cells can differentiate into effector cytotoxic T cells that target pathogens for destruction or memory cells that are ready to respond to subsequent exposures.
As noted, proliferation and differentiation of cytotoxic T cells is also stimulated by cytokines secreted from TH1 cells activated by the same foreign epitope. The co-stimulation that comes from these TH1 cells is provided by secreted cytokines. Although it is possible for activation of cytotoxic T cells to occur without stimulation from TH1 cells, the activation is not as effective or long-lasting.
Once activated, cytotoxic T cells serve as the effector cells of cellular immunity, recognizing and kill cells infected with intracellular pathogens through a mechanism very similar to that of NK cells. However, whereas NK cells recognize nonspecific signals of cell stress or abnormality, cytotoxic T cells recognize infected cells through antigen presentation of pathogen-specific epitopes associated with MHC I. Once an infected cell is recognized, the TCR of the cytotoxic T cell binds to the epitope and releases perforin and granzymes that destroy the infected cell (Figure 8.34). Perforin is a protein that creates pores in the target cell, and granzymes are proteases that enter the pores and induce apoptosis. This mechanism of programmed cell death is a controlled and efficient means of destroying and removing infected cells without releasing the pathogens inside to infect neighboring cells, as might occur if the infected cells were simply lysed.
Part 4: B Lymphocytes and Humoral Immunity
Humoral immunity refers to mechanisms of the adaptive immune defenses that are mediated by antibodies secreted by B lymphocytes, or B cells. This section will focus on B cells and discuss their production and maturation, receptors, and mechanisms of activation.
B Cell Production and Maturation: Like T cells, B cells are formed from multipotent hematopoietic stem cells (HSCs) in the bone marrow and follow a pathway through lymphoid stem cell and lymphoblast (see Figure 8.10). Unlike T cells, however, lymphoblasts destined to become B cells do not leave the bone marrow and travel to the thymus for maturation. Rather, eventual B cells continue to mature in the bone marrow.
The first step of B cell maturation is an assessment of the functionality of their antigen-binding receptors. This occurs through positive selection for B cells with normal functional receptors. A mechanism of negative selection is then used to eliminate self-reacting B cells and minimize the risk of autoimmunity. Negative selection of self-reacting B cells can involve elimination by apoptosis, editing or modification of the receptors so they are no longer self-reactive. Immature B cells that pass the selection in the bone marrow then travel to the spleen for their final stages of maturation. There they become naïve mature B cells, i.e., mature B cells that have not yet been activated.
B-Cell Receptors: Like T cells, B cells possess antigen-specific receptors with diverse specificities. Although they rely on T cells for optimum function, B cells can be activated without help from T cells. B-cell receptors (BCRs) for naïve mature B cells are membrane-bound monomeric forms of IgD and IgM. They have two identical heavy chains and two identical light chains connected by disulfide bonds into a basic “Y” shape. The trunk of the Y-shaped molecule, the constant region of the two heavy chains, spans the B cell membrane. The two antigen-binding sites exposed to the exterior of the B cell are involved in the binding of specific pathogen epitopes to initiate the activation process. It is estimated that each naïve mature B cell has upwards of 100,000 BCRs on its membrane, and each of these BCRs has an identical epitope-binding specificity.
One important difference between BCRs and TCRs is the way they can interact with antigenic epitopes. Whereas TCRs can only interact with antigenic epitopes that are presented within the antigen-binding cleft of MHC I or MHC II, BCRs do not require antigen presentation with MHC; they can interact with epitopes on free antigens or with epitopes displayed on the surface of intact pathogens. Another important difference is that TCRs only recognize protein epitopes, whereas BCRs can recognize epitopes associated with different molecular classes (e.g., proteins, polysaccharides, lipopolysaccharides).
Activation of B cells occurs through different mechanisms depending on the molecular class of the antigen. Activation of a B cell by a protein antigen requires the B cell to function as an APC, presenting the protein epitopes with MHC II to helper T cells. Because of their dependence on T cells for activation of B cells, protein antigens are classified as T-dependent antigens. In contrast, polysaccharides, lipopolysaccharides, and other non-protein antigens are considered T-independent antigens because they can activate B cells without antigen processing and presentation to T cells.
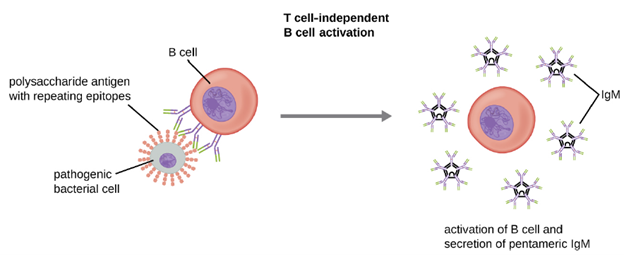
T Cell-Independent Activation of B cells: Activation of B cells without the cooperation of helper T cells is referred to as T cell-independent activation and occurs when BCRs interact with T-independent antigens. T-independent antigens (e.g., polysaccharide capsules, lipopolysaccharide) have repetitive epitope units within their structure, and this repetition allows for the cross-linkageof multiple BCRs, providing the first signal for activation (Figure 8.35). Because T cells are not involved, the second signal has to come from other sources, such as interactions of toll-like receptors with PAMPs or interactions with factors from the complement system.
Once a B cell is activated, it undergoes clonal proliferation and daughter cells differentiate into plasma cells. Plasma cells are antibody factories that secrete large quantities of antibodies. After differentiation, the surface BCRs disappear and the plasma cell secretes pentameric IgM molecules that have the same antigen specificity as the BCRs (Figure 8.35). The T cell-independent response is short-lived and does not result in the production of memory B cells. Thus it will not result in a secondary response to subsequent exposures to T-independent antigens.
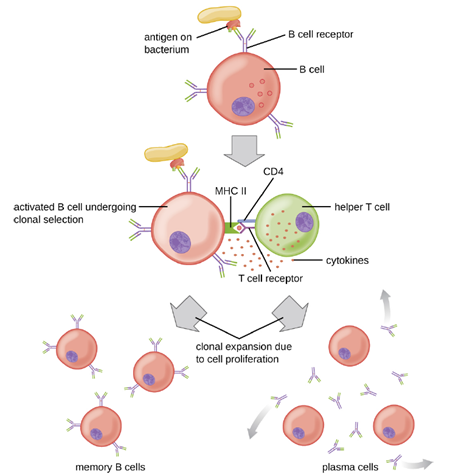
T Cell-Dependent Activation of B cells: T cell-dependent activation of B cells is more complex than T cell-independent activation, but the resulting immune response is stronger and develops memory. T cell-dependent activation can occur either in response to free protein antigens or to protein antigens associated with an intact pathogen. Interaction between the BCRs on a naïve mature B cell and a free protein antigen stimulate internalization of the antigen, whereas interaction with antigens associated with an intact pathogen initiates the extraction of the antigen from the pathogen before internalization. Once internalized inside the B cell, the protein antigen is processed and presented with MHC II. The presented antigen is then recognized by helper T cells specific to the same antigen. The TCR of the helper T cell recognizes the foreign antigen, and the T cell’s CD4 molecule interacts with MHC II on the B cell. The coordination between B cells and helper T cells that are specific to the same antigen is referred to as linked recognition.
Once activated by linked recognition, TH2 cells produce and secrete cytokines that activate the B cell and cause proliferation into clonal daughter cells. After several rounds of proliferation, additional cytokines provided by the TH2 cells stimulate the differentiation of activated B cell clones into memory B cells, which will quickly respond to subsequent exposures to the same protein epitope, and plasma cells that lose their membrane BCRs and initially secrete pentameric IgM (Figure 8.36).
After initial secretion of IgM, cytokines secreted by TH2 cells stimulate the plasma cells to switch from IgM production to production of IgG, IgA, or IgE. This process, called class switching or isotype switching, allows plasma cells cloned from the same activated B cell to produce a variety of antibody classes with the same epitope specificity. Class switching is accomplished by genetic rearrangement of gene segments encoding the constant region, which determines an antibody’s class. The variable region is not changed, so the new class of antibody retains the original epitope specificity.
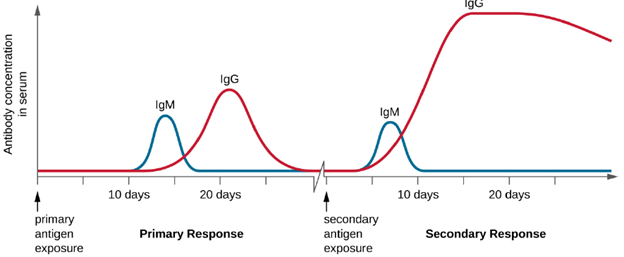
Primary and Secondary Responses: T cell-dependent activation of B cells plays an important role in both the primary and secondary responses associated with adaptive immunity. With the first exposure to a protein antigen, a T cell-dependent primary antibody response occurs. The initial stage of the primary response is a lag period, or latent period, of approximately 10 days, during which no antibody can be detected in serum. This lag period is the time required for all of the steps of the primary response, including naïve mature B cell binding of antigen with BCRs, antigen processing and presentation, helper T cell activation, B cell activation, and clonal proliferation. The end of the lag period is characterized by a rise in IgM levels in the serum, as TH2 cells stimulate B cell differentiation into plasma cells. IgM levels reach their peak around 14 days after primary antigen exposure; at about this same time, TH2 stimulates antibody class switching, and IgM levels in serum begin to decline. Meanwhile, levels of IgG increase until they reach a peak about three weeks into the primary response (Figure 8.37).
During the primary response, some of the cloned B cells are differentiated into memory B cells programmed to respond to subsequent exposures. This secondary response occurs more quickly and forcefully than the primary response. The lag period is decreased to only a few days and the production of IgG is significantly higher than observed for the primary response (Figure 8.37). In addition, the antibodies produced during the secondary response are more effective and bind with higher affinity to the targeted epitopes. Plasma cells produced during secondary responses live longer than those produced during the primary response, so levels of specific antibody remain elevated for a longer period of time.
For many diseases, prevention is the best form of treatment, and few strategies for disease prevention are as effective as vaccination. Vaccination is a form of artificial immunity. By artificially stimulating the adaptive immune defenses, a vaccine triggers memory cell production similar to that which would occur during a primary response. In so doing, the patient is able to mount a strong secondary response upon exposure to the pathogen—but without having to first suffer through an initial infection. In this section, we will explore several different kinds of artificial immunity along with various types of vaccines and the mechanisms by which they induce artificial immunity.
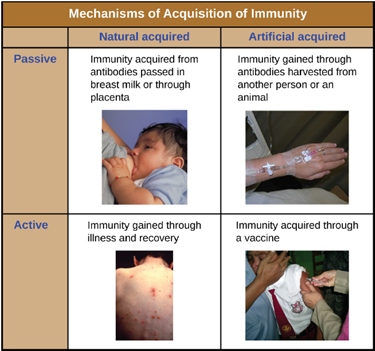
Classifications of Adaptive Immunity: All forms of adaptive immunity can be described as either active or passive. Active immunity refers to the activation of an individual’s own adaptive immune defenses, whereas passive immunity refers to the transfer of adaptive immune defenses from another individual or animal. Active and passive immunity can be further subdivided based on whether the protection is acquired naturally or artificially.
Natural active immunity is adaptive immunity that develops after natural exposure to a pathogen (Figure 8.38). Examples would include the lifelong immunity that develops after recovery from a chickenpox or measles infection (although an acute infection is not always necessary to activate adaptive immunity). The length of time that an individual is protected can vary substantially depending upon the pathogen and antigens involved. For example, activation of adaptive immunity by protein spike structures during an intracellular viral infection can activate lifelong immunity, whereas activation by carbohydrate capsule antigens during an extracellular bacterial infection may activate shorter-term immunity.
Natural passive immunity involves the natural passage of antibodies from a mother to her child before and after birth. IgG is the only antibody class that can cross the placenta from mother’s blood to the fetal blood supply. Placental transfer of IgG is an important passive immune defense for the infant, lasting up to six months after birth. Secretory IgA can also be transferred from mother to infant through breast milk.
Artificial passive immunity refers to the transfer of antibodies produced by a donor (human or animal) to another individual. This transfer of antibodies may be done as a prophylactic measure (i.e., to prevent disease after exposure to a pathogen) or as a strategy for treating an active infection. For example, artificial passive immunity is commonly used for post-exposure prophylaxis against rabies, hepatitis A, hepatitis B, and chickenpox (in high risk individuals). Active infections treated by artificial passive immunity include cytomegalovirus infections in immunocompromised patients and Ebola virus infections. In 1995, eight patients in the Democratic Republic of the Congo with active Ebola infections were treated with blood transfusions from patients who were recovering from Ebola. Only one of the eight patients died (a 12.5% mortality rate), which was much lower than the expected 80% mortality rate for Ebola in untreated patients. Artificial passive immunity is also used for the treatment of diseases caused by bacterial toxins, including tetanus, botulism, and diphtheria.
Artificial active immunity is the foundation for vaccination. It involves the activation of adaptive immunity through the deliberate exposure of an individual to weakened or inactivated pathogens, or preparations consisting of key pathogen antigens.
Herd Immunity: The four kinds of immunity just described result from an individual’s adaptive immune system. For any given disease, an individual may be considered immune or susceptible depending on his or her ability to mount an effective immune response upon exposure. Thus, any given population is likely to have some individuals who are immune and other individuals who are susceptible. If a population has very few susceptible individuals, even those susceptible individuals will be protected by a phenomenon called herd immunity. Herd immunity has nothing to do with an individual’s ability to mount an effective immune response; rather, it occurs because there are too few susceptible individuals in a population for the disease to spread effectively.
Vaccination programs create herd immunity by greatly reducing the number of susceptible individuals in a population. Even if some individuals in the population are not vaccinated, as long as a certain percentage is immune (either naturally or artificially), the few susceptible individuals are unlikely to be exposed to the pathogen. However, because new individuals are constantly entering populations (for example, through birth or relocation), vaccination programs are necessary to maintain herd immunity.
Vaccination: The English physician Edward Jenner (1749–1823) is generally credited with developing the modern process of vaccination. Jenner observed that milkmaids who developed cowpox, a disease similar to smallpox but milder, were immune to the more serious smallpox. This led Jenner to hypothesize that exposure to a less virulent pathogen could provide immune protection against a more virulent pathogen. In 1796, Jenner tested his hypothesis by obtaining infectious samples from a milkmaid’s active cowpox lesion and injecting the materials into a young boy. The boy developed a mild infection that included a low-grade fever, discomfort in his axillae (armpit) and loss of appetite. When the boy was later infected with infectious samples from smallpox lesions, he did not contract smallpox.[4] This new approach was termed vaccination, a name deriving from the use of cowpox (Latin vacca meaning “cow”) to protect against smallpox. Today, we know that Jenner’s vaccine worked because the cowpox virus is genetically and antigenically related to the Variola viruses that caused smallpox. Exposure to cowpox antigens resulted in a primary response and the production of memory cells that identical or related epitopes of Variola virus upon a later exposure to smallpox.
The success of Jenner’s smallpox vaccination led other scientists to develop vaccines for other diseases. Perhaps the most notable was Louis Pasteur, who developed vaccines for rabies, cholera, and anthrax. During the 20th and 21st centuries, effective vaccines were developed to prevent a wide range of diseases caused by viruses (e.g., chickenpox and shingles, hepatitis, measles, mumps, polio, and yellow fever) and bacteria (e.g., diphtheria, pneumococcal pneumonia, tetanus, and whooping cough).
Classes of Vaccines: For a vaccine to provide protection against a disease, it must expose an individual to pathogen-specific antigens that will stimulate a protective adaptive immune response. By its very nature, this entails some risk. As with any pharmaceutical drug, vaccines have the potential to cause adverse effects. However, the ideal vaccine causes no severe adverse effects and poses no risk of contracting the disease that it is intended to prevent. Various types of vaccines have been developed with these goals in mind. These different classes of vaccines are described in the next section and summarized in Table 8.6.
- Live Attenuated Vaccines: Live attenuated vaccines expose an individual to a weakened strain of a pathogen with the goal of establishing a subclinical infection that will activate the adaptive immune defenses. Pathogens are attenuated to decrease their virulence using methods such as genetic manipulation (to eliminate key virulence factors) or long-term culturing in an unnatural host or environment (to promote mutations and decrease virulence).
By establishing an active infection, live attenuated vaccines stimulate a more comprehensive immune response than some other types of vaccines. Live attenuated vaccines activate both cellular and humoral immunity and stimulate the development of memory for long-lasting immunity. In some cases, vaccination of one individual with a live attenuated pathogen can even lead to natural transmission of the attenuated pathogen to other individuals. This can cause the other individuals to also develop an active, subclinical infection that activates their adaptive immune defenses.
Disadvantages associated with live attenuated vaccines include the challenges associated with long-term storage and transport as well as the potential for a patient to develop signs and symptoms of disease during the active infection (particularly in immunocompromised patients). There is also a risk of the attenuated pathogen reverting back to full virulence. Table 8.6 lists examples live attenuated vaccines.
- Inactivated Vaccines: Inactivated vaccines contain whole pathogens that have been killed or inactivated with heat, chemicals, or radiation. For inactivated vaccines to be effective, the inactivation process must not affect the structure of key antigens on the pathogen.
Because the pathogen is killed or inactive, inactivated vaccines do not produce an active infection, and the resulting immune response is weaker and less comprehensive than that provoked by a live attenuated vaccine. Typically the response involves only humoral immunity, and the pathogen cannot be transmitted to other individuals. In addition, inactivated vaccines usually require higher doses and multiple boosters, possibly causing inflammatory reactions at the site of injection.
Despite these disadvantages, inactivated vaccines do have the advantages of long-term storage stability and ease of transport. Also, there is no risk of causing severe active infections. However, inactivated vaccines are not without their side effects. Table 8.6 lists examples of inactivated vaccines.
- Subunit Vaccines: Whereas live attenuated and inactive vaccines expose an individual to a weakened or dead pathogen, subunit vaccines only expose the patient to the key antigens of a pathogen—not whole cells or viruses. Subunit vaccines can be produced either by chemically degrading a pathogen and isolating its key antigens or by producing the antigens through genetic engineering. Because these vaccines contain only the essential antigens of a pathogen, the risk of side effects is relatively low. Table 8.6 lists examples of subunit vaccines.
- Toxoid Vaccines: Like subunit vaccines, toxoid vaccines do not introduce a whole pathogen to the patient; they contain inactivated bacterial toxins, called toxoids. Toxoid vaccines are used to prevent diseases in which bacterial toxins play an important role in pathogenesis. These vaccines activate humoral immunity that neutralizes the toxins. Table 8.6 lists examples of toxoid vaccines.
- Conjugate Vaccines: A conjugate vaccine is a type of subunit vaccine that consists of a protein conjugated to a capsule polysaccharide. Conjugate vaccines have been developed to enhance the efficacy of subunit vaccines against pathogens that have protective polysaccharide capsules that help them evade phagocytosis, causing invasive infections that can lead to meningitis and other serious conditions. The subunit vaccines against these pathogens introduce T-independent capsular polysaccharide antigens that result in the production of antibodies that can opsonize the capsule and thus combat the infection; however, children under the age of two years do not respond effectively to these vaccines. Children do respond effectively when vaccinated with the conjugate vaccine, in which a protein with T-dependent antigens is conjugated to the capsule polysaccharide. The conjugated protein-polysaccharide antigen stimulates production of antibodies against both the protein and the capsule polysaccharide. Table 8.6 lists examples of conjugate vaccines.
- Nucleic acid Vaccines: Nucleic acid (DNA) vaccines are one of the newest vaccine types. In this vaccine, plasmids of DNA are injected into the patient. These plasmids are taken up by cells and used at a template to create the encoded pathogen proteins. These proteins then stimulate an immune response. The advantages of these vaccines are they are cheap and easy to produce, since you do not need to produce any pathogens, and produce strong immune responses. However, it is not possible to produce polysaccharides this way, so vaccines against capsules are not possible. Previously, only vaccines for animals have been approved, such as a horse vaccine for West Nile virus, but the recent Zika virus outbreak has resulted in the development a DNA vaccine for use in humans.
Table 8.6: Classes of Vaccines
| Class | Description | Advantages | Disadvantages | Examples |
| Live attenuated | Weakened strain of whole pathogen | Cellular and humoral immunity
Long-lasting immunity |
Difficult to store and transport
Risk of infection in immunocompromised patients |
Chickenpox, German measles, measles, mumps, tuberculosis, typhoid fever, yellow fever |
| Inactivated | Whole pathogen killed or inactivated with heat, chemicals, or radiation | Ease of storage and transport
No risk of severe active infection |
Weaker immunity (humoral only)
Higher doses and more boosters required |
Cholera, hepatitis A, influenza, plague, rabies |
| Subunit | Immunogenic antigens | Lower risk of side effects | Limited longevity
Multiple doses required No protection against antigenic variation |
Anthrax, hepatitis B, influenza, meningitis, papillomavirus, pneumococcal pneumonia, whooping cough |
| Toxoid | Inactivated bacterial toxin | Humoral immunity to neutralize toxin | Does not prevent infection | Botulism, diphtheria, pertussis, tetanus |
| Conjugate | Capsule polysaccharide conjugated to protein | T-dependent response to capsule
Better response in young children |
Costly to produce
No protection against antigenic variation May interfere with other vaccines
|
Meningitis (Haemophilus influenzae, Streptococcus pneumoniae, Neisseria meningitides) |
| Nucleic acid | DNA Plasmid | Cellular and humoral immunity
Cheap to produce |
Not possible to target polysaccharides | West Nile (horse), Zika |
