3 Bioinformatics assignment 1
Pardis Mani and kathleef
Contents:
Why do we want to do this assignment?
What is happening in each step?
1. Use BLAST search to find DNA or protein sequences that closely resemble your sequence
1.1. First, access the BLASTN tool
1.2. Input your known gene sequence
1.4. Run BLAST & analyze the results
1.5. We can also observe the graphic view of the gene
Common Mistakes & Troubleshooting
2. Identifying the longest Open Reading Frame (ORF) using ExPASy
2.1. Access the translation tool
2.4. Analyze the six reading frames output
2.5. Click on the first Methionine (M) in the longest ORF
Common Mistakes & Troubleshooting
3. Analyzing the predicted protein sequence using BLASTP
3.2. Input your protein sequence
Common Mistakes & Troubleshooting
Why do we want to do this project?
Western Red Cedar (WRC) is common in the coastal and interior rainforests of the Pacific Northwest of America (Gonzalez et al. 1997). WRC is a 1-billion-dollar industry in British Columbia, given its low cut volume (Gregory et al. 2018). Heartwood is the primary component of the WRC used in industry, and understanding the genes essential for heartwood formation can be highly beneficial to industries. To identify these genes, a series of uniquely expressed or highly upregulated mRNAs in this part of the wood was isolated and converted into a cDNA library. If a gene is only expressed in one part of an organism, or if it’s explicitly up-regulated in that part, it is a good clue for us to suspect that the resulting protein has a function in forming something in that part! [They use RNA seq technique to find which genes’ mRNAs to choose, but how they did this is out of the scope of this course, so don’t worry about it.]
What do we want to do?
Each pair of students was assigned two cDNA sequences from this cDNA library. We want to compare the sequence of these cDNAs and see if they function in heartwood formation in WRC.
Let’s get familiar with a process in genetics/genomics known as gene annotation.
Sometimes, we try to guess the function of a gene based on the similarity of its sequence with the sequence of another gene for which we already know its function. Since these two genes have very similar sequences and, therefore, will result in similar protein products, they might have similar functions in the cell. This process is known as “annotating” the gene function.
So, our end goal in this assignment is to attempt to evaluate the proposed approximate functions of the two designated cDNAs.
What steps are involved?
To do this, there are some steps to take:
-
-
- Use our known gene sequence to find all similar gene sequences. We use BLASTN for this step.
- Use our known gene sequence to find the longest open reading frame (ORF) and therefore, the protein sequence. We use Expasy for this step.
- Use the protein sequence obtained from the previous step to determine the function of your resulting protein. We employ BLASTP for this step.
- Use this information to talk about the function of your gene :).
-
What is happening in each step?
Now let’s talk about each step in more depth:
1. Use BLAST search to find DNA or protein sequences that closely resemble your sequence:
The process of matching nucleotides from two DNA/ amino acid sequences to identify the most similar nucleotide/ amino acid sequence to our sequence of interest is called alignment (Fassler and Cooper 2011). This is where the name BLAST originates; the Basic Local Alignment Search Tool (Altschul et al. 1990) is a method for aligning our sequence of interest with known sequences, establishing a threshold for similarity and sensitivity in our search. We set the sensitivity of BLAST through the settings on the searching webpage, and we select the degree of similarity we desire among the results BLAST provides.
1.1. First, access the BLASTN tool:
-
-
- BLASTN compares a DNA sequence with other DNA sequences in the database.
- Go to the NCBI BLAST website.
- Click on “Nucleotide BLAST (BLASTN)” since we are working with cDNA sequences.
-
1.2. Input your known gene sequence:
-
-
- Copy and paste your cDNA sequence into the query box.
- Ensure the sequence is formatted properly:
-
>Sample_cDNA
ATGCGTACGTTGCTAGCTAGGCT…
(Include the > symbol before the sequence name.)
1.3. Leave the database setting as default (it should already be set to “Standard databases (nr etc.)”):
-
-
- This is where we set the sensitivity of our search. First, we try to find the most similar sequences. If we can’t find any results, we move on to search for sequences with lower similarities:
- Optimize for Similar Sequences: Use the “somewhat similar sequences” setting if no results appear.
-
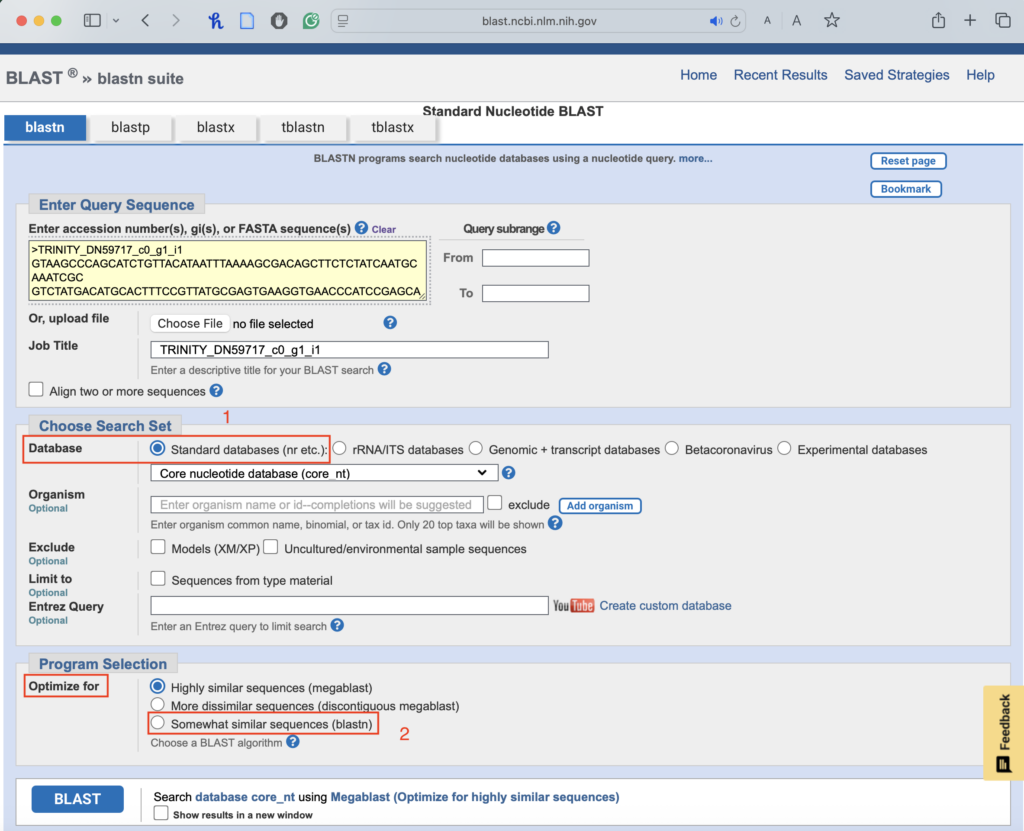
1.4. Run BLAST & analyze the results:
-
-
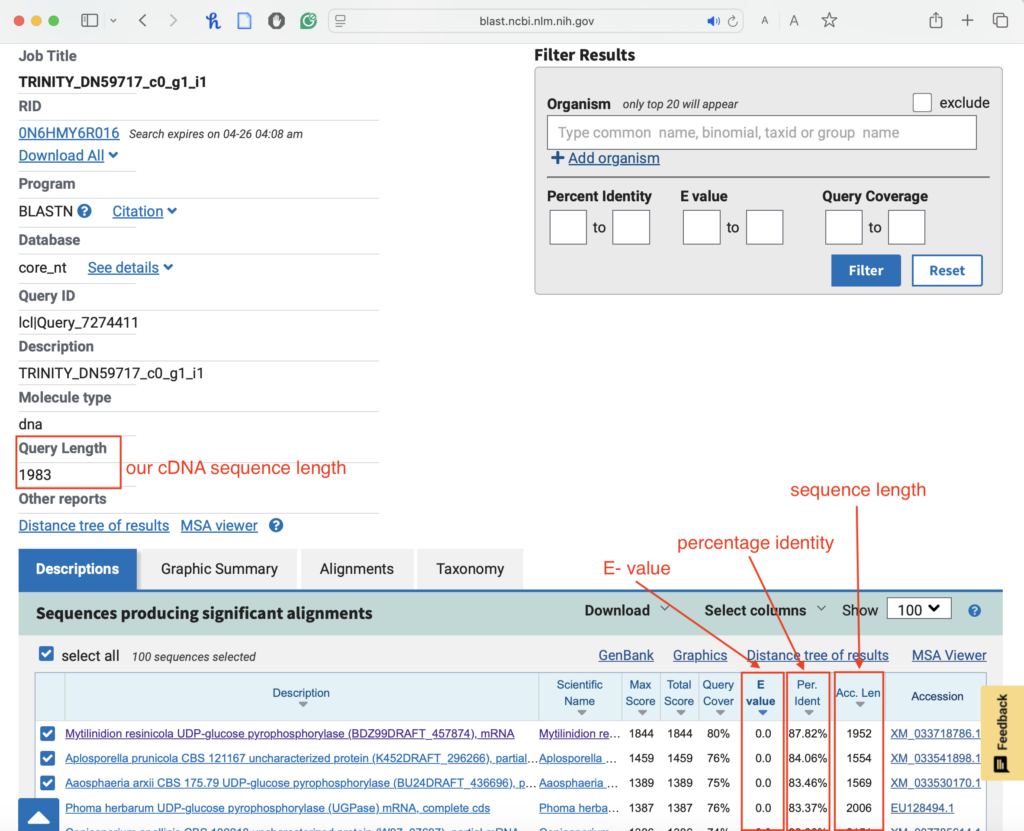
- Now we choose the similarity threshold. To do so, BLAST provides us with some values:
- E value: The Expectation value, or Expect value, indicates how many alignments we expect to find in the database by chance that match our sequence equally or better than the specified threshold (Fassler and Cooper 2011). An excellent e-value is below 1e-180, which exceeds the computer’s processing capabilities, and is therefore represented as 0.0.
- Percentage identity: The extent to which two sequences have identical residues at corresponding positions (Fassler and Cooper 2011).
- Identify the top hits based on E values (should be < 0.05)
- Examine the alignment–the best matches should have a high percentage identity and similar sequence length.
- The lengths of the query and subject sequences should not differ by more than 20%.
- Now we choose the similarity threshold. To do so, BLAST provides us with some values:
-
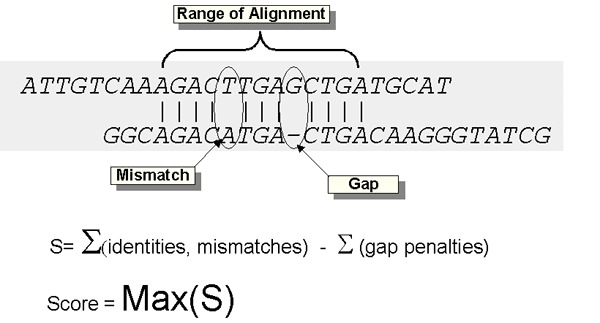
- The BLAST scoring system uses matrices to award points for matches between nucleotides and penalize mismatches and gaps (insertions/deletions).
- Matches: Identical nucleotides between the query (your sequence) and the subject (database sequence) receive positive scores.
- Mismatches: Differences between nucleotides result in minor penalties.
- Gaps (insertions/deletions): BLAST introduces gaps to optimize alignment, but longer gaps receive higher penalties.
- The sum of these factors determines the sequence similarity.
-
-
- The results are organized with the highest matches listed first, showcasing the most similar sequences with minimal mismatches or gaps.
- E-values depend on the query sequence length and the database size, indicating that even perfect matches with short sequences may result in low e-values.
- A minimum of 30% of amino acids must be identical.
-
1.5. We can also observe the graphic view of the gene:
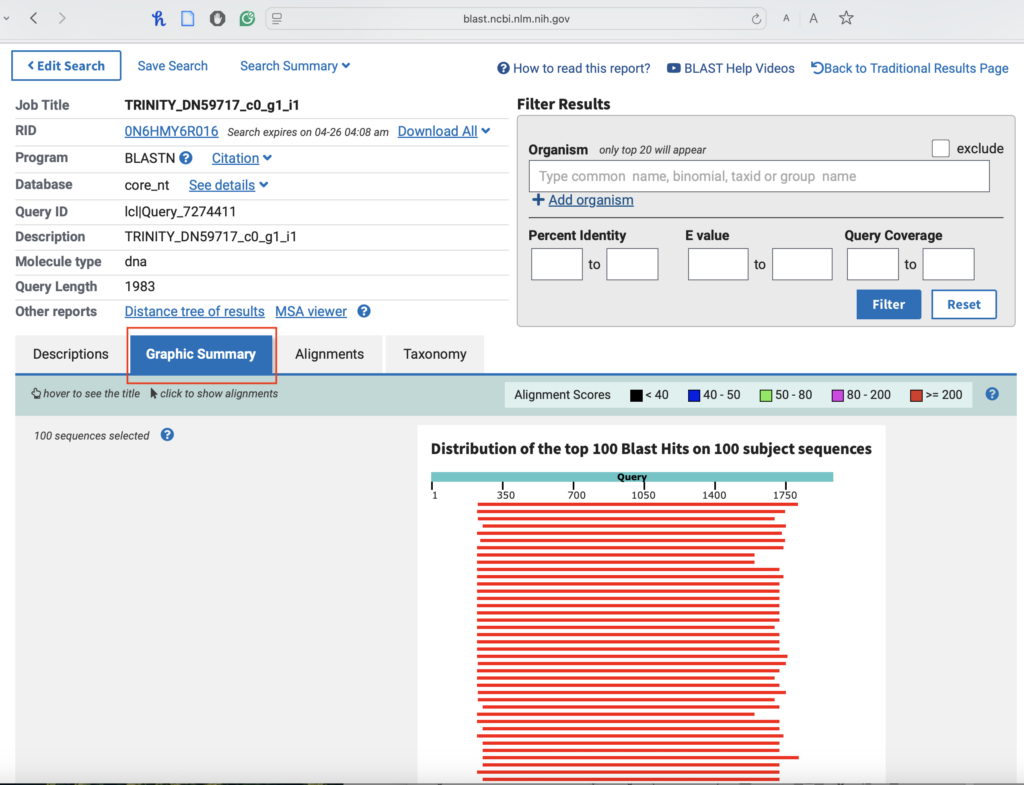
Common Mistakes & Troubleshooting:
-
-
-
- No significant hits found? Try using the “somewhat similar sequences” setting.
- Low identity or poor alignment? Ensure that the entire sequence was pasted accurately (including all bases)
- Confusing E-values? Keep in mind: Smaller E-values (e.g., 1e-50 or smaller) indicate strong matches, while values above 0.05 suggest random similarity.
-
-
2. Identifying the longest Open Reading Frame (ORF) using ExPASy
Many amino acids bind together to form proteins. There are 20 distinct amino acids encoded by combinations of three nucleic acids known as codons. Since four different nucleotides comprise DNA (A, T, C, G), there are 64 possible codons [combination of 4 x 4 x 4 = 64]. Therefore, most amino acids have more than one possible codon. Protein synthesis begins at a start codon and ends at a stop codon. There are three possible stop codons: UAA, UAG, and UGA, corresponding to TAA, TAG, and TGA in the DNA.
Start codons are a bit more complicated as they can appear in the middle of genes as well as at the start:
In eukaryotes, the start codon is always ATG, which designates Methionine (Met) as the first amino acid in all proteins, though it is often immediately removed. Methionine can be present at any position within the protein.
In prokaryotes, the start codons can be ATG, GTG, or TTG. Although ATG is the most common, approximately 30% of genes begin with GTG or TTG.
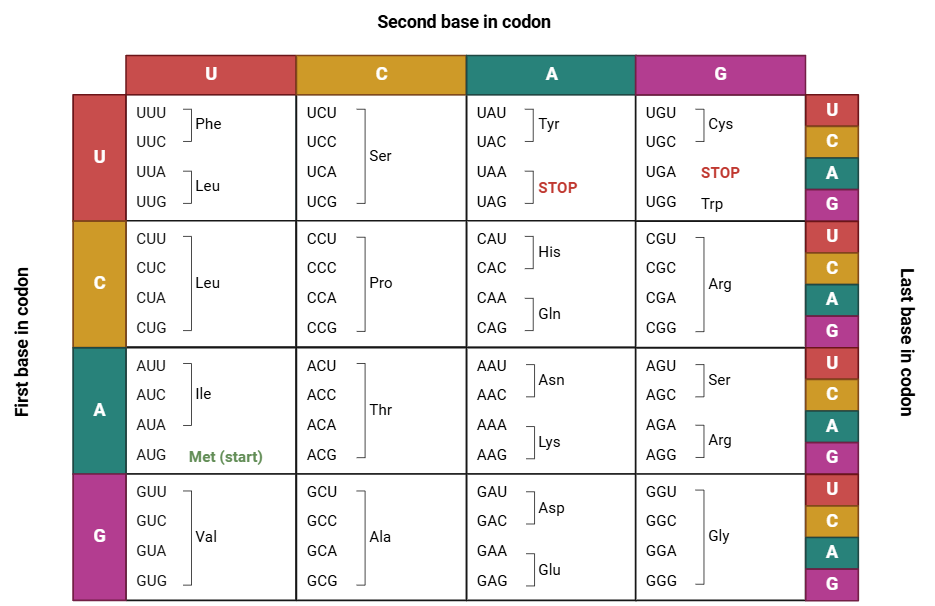
Codons comprise three bases, allowing for three possible “reading frames” on an RNA (or DNA) strand. This depends on whether the reading begins from the first, second, or third base. Each reading frame results in distinct proteins. A gene utilizes only one reading frame, so once the ribosome begins, it counts groups of three bases to synthesize the corresponding protein.
2.1. Access the translation tool:
-
-
- Go to the ExPASy Translate Tool.
-
2.2. Input your cDNA sequence:
-
-
- Paste the same cDNA sequence utilized in your BLASTN search into the input box.
- Ensure the sequence is clean and free of any non-DNA characters.
-
2.3. Use the default settings:
-
-
- Keep “Standard” genetic code selected.
- For “Output Format,” keep “Compact: M, -, no spaces.”
- Press “Translate.”
-
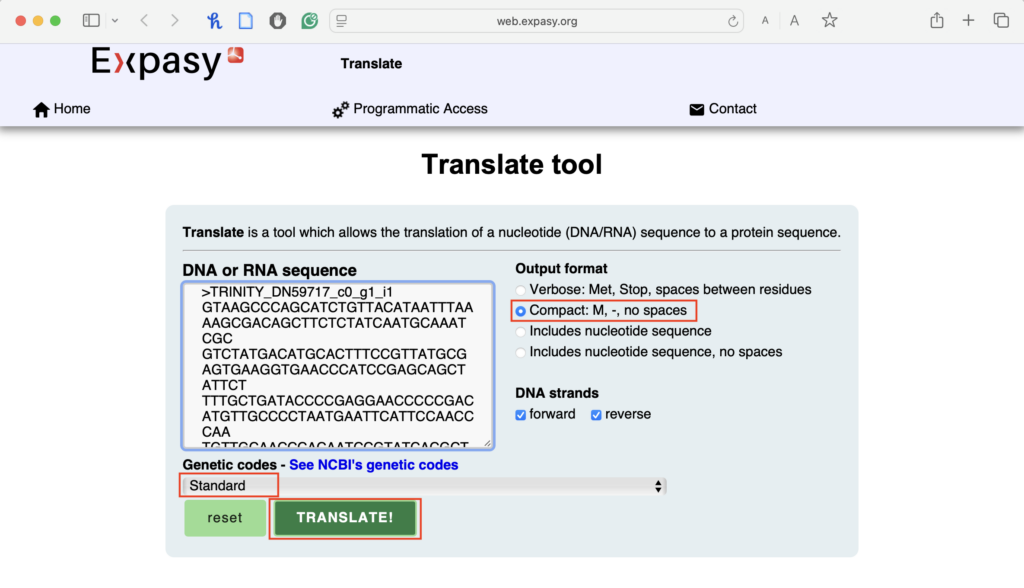
2.4. Analyze the six reading frames output:
-
-
- You will see six frames: three from the forward strand and three from the reverse complement.
- Look for the longest stretch of amino acids without stop codons (stop codons are specified with “-“).
- A valid ORF starts with M (Methionine) and ends at a stop codon. In this tool, ORF is highlighted in red for easier recognition.
- The longest valid ORF is typically the correct coding region.
-
In our current example, these are the six reading frames:


2.5. Click on the first Methionine (M) in the longest ORF:
-
-
- This will open the Fasta format of your amino acid sequence.
- You can either copy the Fasta format of this protein sequence or download it. You will use this amino acid sequence in the next step for BLASTP.
-
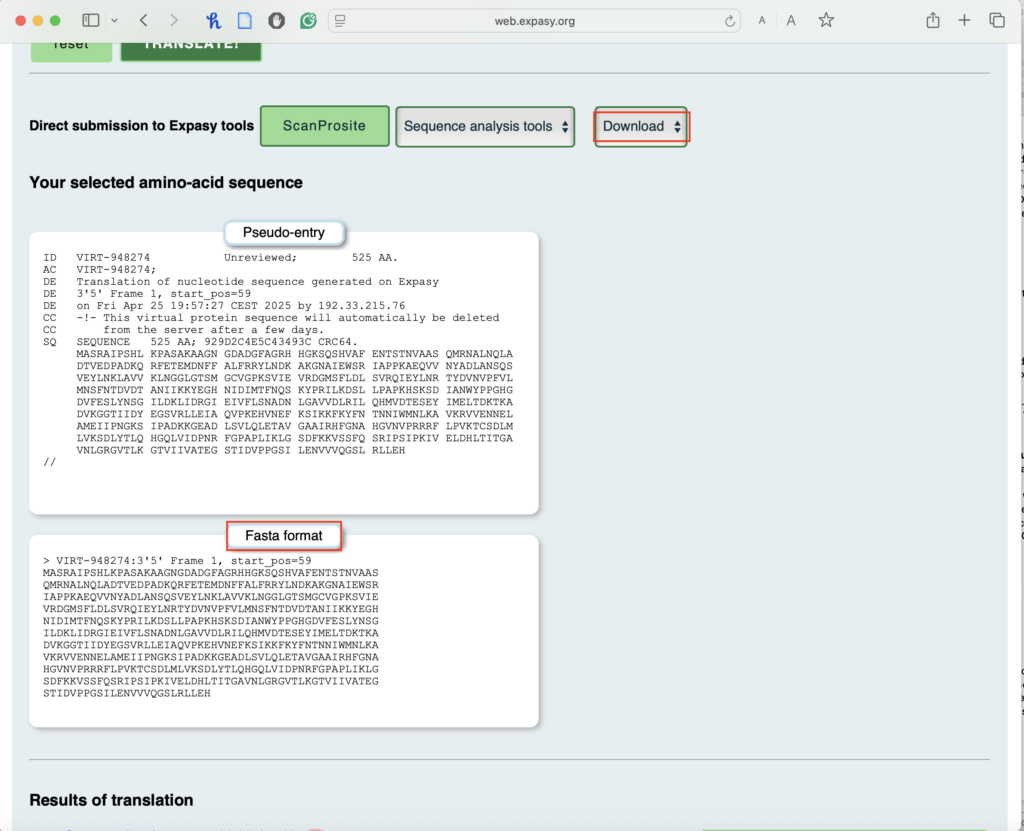
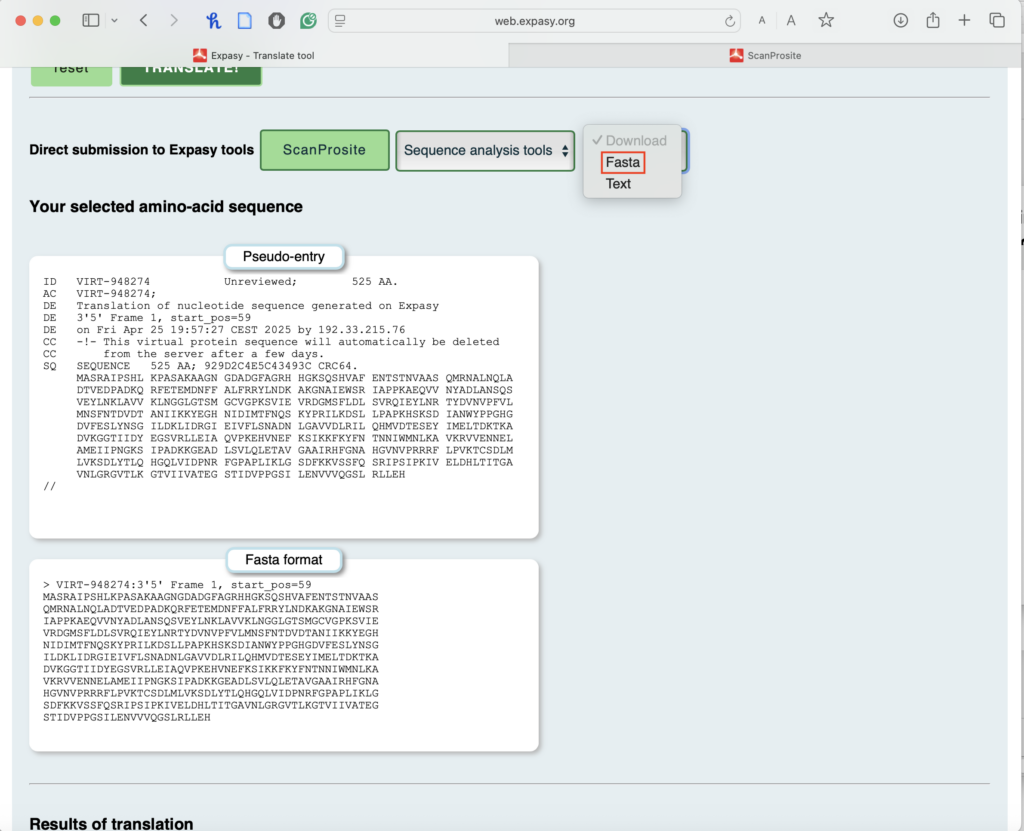
This is what your downloaded file looks like:
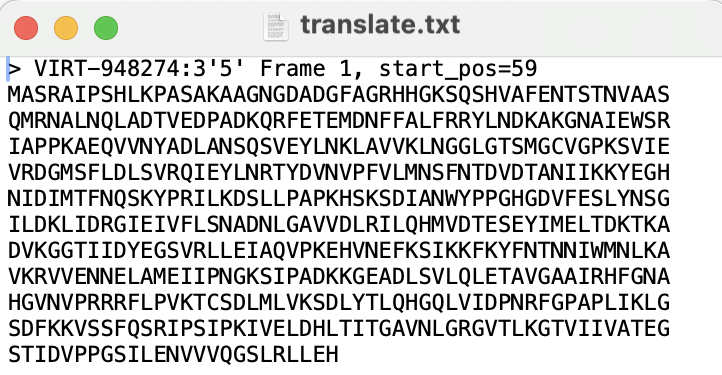
-
-
- For “Output Format,” choose “Includes nucleotide sequence” to match amino acids with their DNA positions.
- Press “Translate Sequence.”
-

This is what the result looks like:

Some part of the ORF have been deleted to save space (you can do the same in your report).

Common Mistakes & Troubleshooting:
-
-
-
-
Choose a short ORF? Always select the longest ORF that starts with a Methionine (M) and ends with a stop codon (-).
-
Mistook an internal Methionine (M) for the start codon? Start translation at the first Methionine (M) of the longest continuous amino acid sequence. However, this is less of a problem in our case, as ExPASy highlights the ORF.
-
Forgot to check reverse frames? Make sure you review all six frames (three forward, three reverse) when looking for the longest ORF.
-
Pasted an incomplete sequence? Double-check that you paste the full cDNA sequence into ExPASy.
-
Misread stop codons? Remember that the “-” symbol in ExPASy output indicates a stop codon; your ORF should not contain any “-” until the very end.
-
Assumed the first frame shown is correct? Don’t assume; the correct frame has the longest, uninterrupted stretch, not just the first one listed.
-
Chose a long sequence without a start codon? Ensure the ORF has both a start codon (ATG/M) and a stop codon.
-
-
-
3. Analyzing the predicted protein sequence using BLASTP.
We typically conduct a BLASTP search to determine whether our amino acid sequence matches the DNA sequence’s gene and protein family. If not, you have identified the incorrect ORF. Therefore, in this step, you compare the predicted protein sequence (from the longest ORF) against a protein database to identify similar proteins and infer the function of your gene.
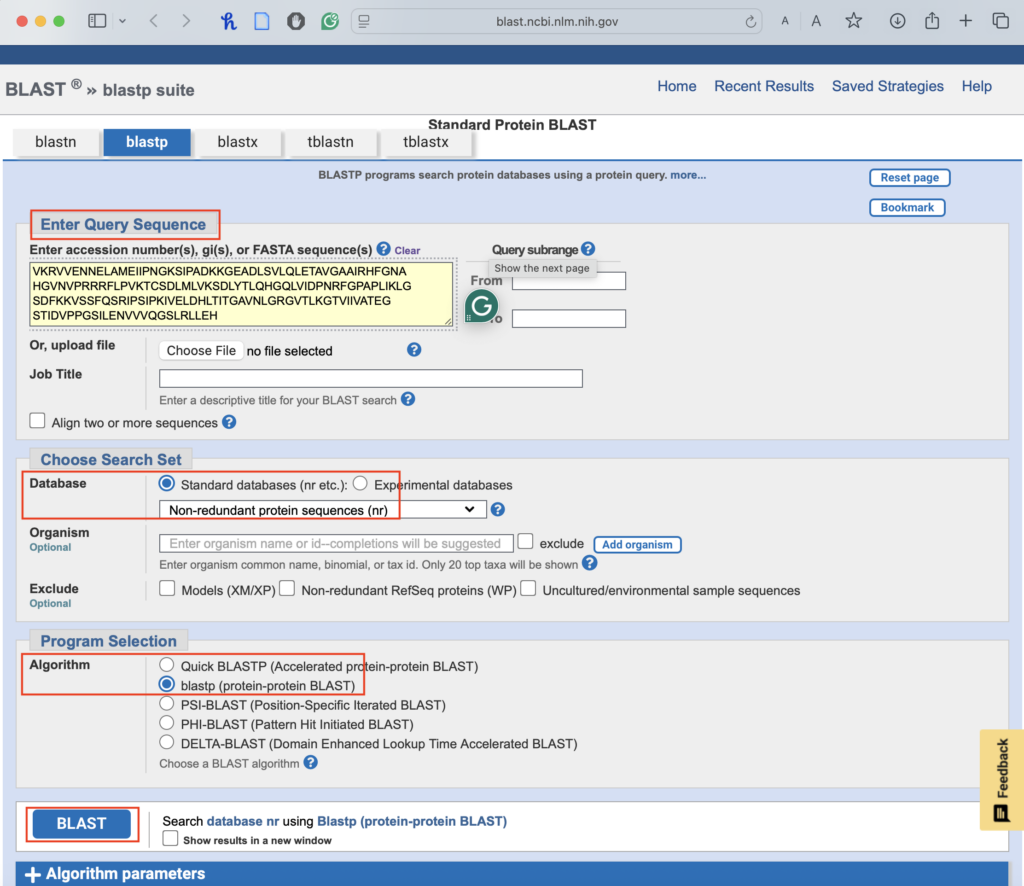
3.1. Access the BLASTP Tool:
-
-
- Go to NCBI BLASTP.
-
3.2. Input your protein sequence:
-
-
- Paste the protein sequence you obtained from ExPASy into the query box.
-
3.3. Use the default settings:
-
-
- Database: Should be preset to “Non-redundant protein sequences (nr)”.
- Algorithm: Keep the default “blastp (protein-protein BLAST)”
- Do not change parameters unless necessary.
-
3.4. Run the search:
-
-
- Click “BLAST” and wait for the results.
-
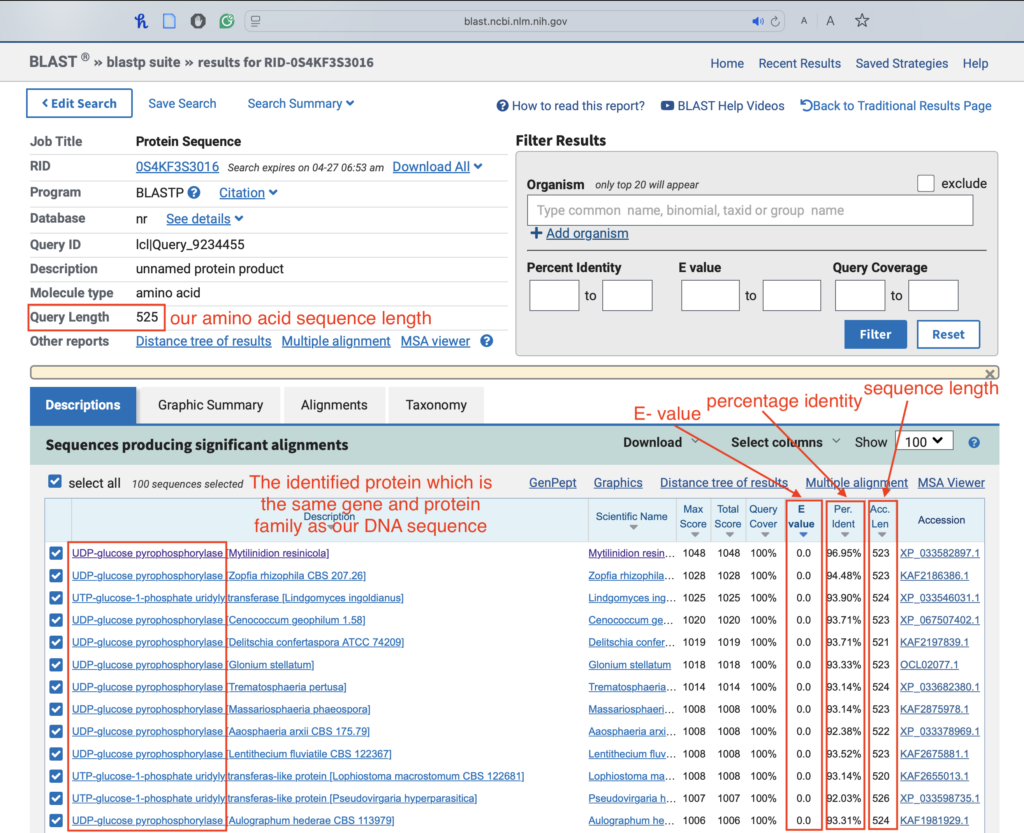
3.5. Interpret the results:
-
-
- Focus on top hits with E-values < 0.05 (strong matches).
- Look at the percentage identity and alignment length:
- Ideally, ≥ 30% identity.
- Query and subject protein lengths should be within ~20% of each other.
-
Choose hits that describe a clear molecular function. Avoid “hypothetical protein” or “unknown function”.
-
Common Mistakes & Troubleshooting:
-
-
-
-
Found only hypothetical proteins or unknown annotations? Scroll down to find matches with specific functional descriptions.
-
E-values are too high (>0.05)? Ensure that you’re using the complete protein sequence from the longest ORF.
-
Top matches have low identity? Reassess if you chose the right ORF and started at the first Methionine (M).
-
-
-
4. Annotating the likely function of the gene.
Read the annotations of top hits to determine the likely function of your protein. Summarize in 3–5 sentences what the likely function of your gene is. Mention both the evidence (BLAST matches, E-values, identity %) and the proposed function.
In our previous example:
>TRINITY_DN59717_c0_g1_i1
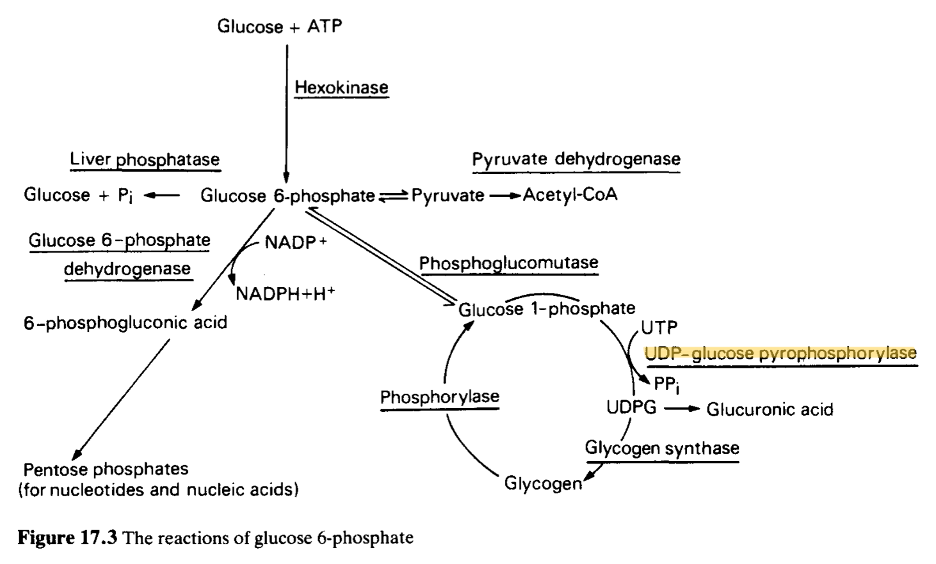
This cDNA was analyzed using BLASTN, which produced many statistically significant alignments to DNA responsible for coding UDP-glucose pyrophosphorylase. The lowest E-value produced was 0.0 by BLASTN. The longest ORF was identified at 3’5’ Frame 1 by ExPasy. We concluded that the correct ORF was identified, as BLASTP produced statistically significant alignment to DNA sequences encoding UDP-glucose pyrophosphorylase. The lowest E-value produced by BLASTP was 0.0. Considering our analysis’s evidence, we conclude that this cDNA encodes a UDP-glucose pyrophosphorylase protein.
The protein was identified as UDP-glucose pyrophosphorylase. It’s essential for glycogenesis, specifically to make UDP-glucose, a key precursor in the glycogen synthesis cycle. In this way, this protein plays an indirect role in energy storage and cell structure maintenance.

References:
Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and D.J. Lipman, 1990 Basic local alignment search tool. Journal of Molecular Biology. 215: 403–410
Aseem, A., 2024 Codon Chart and Codon Wheel – Explained [Accessed 2025-04-24] https://www.biotechreality.com/2024/11/codon-chart-and-codon-wheel-explained.html
Cole, A.S., and J.E. Eastoe, 1988 Carbohydrate metabolism. In: Biochemistry and Oral Biology. Elsevier, 224–248
Fassler, J., and P. Cooper, 2011 BLAST Glossary. In: BLAST® Help [Internet]. National Center for Biotechnology Information (US),
Gonzalez, J.S., Forintek Canada Corp, and Western Red Cedar Lumber Association, 1997 Growth, Properties and Uses of Western Redcedar (Thuja Plicata Donn Ex D. Don.). Forintek Canada Corporation.
Gregory, Christopher., McBeath, Alec., Filipescu, Cosmin., and Canadian Electronic Library (Firm) distributor., 2018 An Economic Assessment of the Western Redcedar Industry in British Columbia. Natural Resources Canada, Victoria, B.C.