10 Investigating Gene Function Part 3 – Knock-down and Knock-out
RNAi and CRISPR-Cas9
Introduction
In this section we will look at ways to go beyond detecting when and where a gene is expressed; we will explore ways of reducing or eliminating the gene’s expression, and using the resulting phenotype to deduce the likely role of the gene product. Understanding when and where the gene is expressed (discussed in Chapter 7 and Chapter 8) will help us know where we should expect to see the phenotype resulting from reduced or no expression of the gene. It is an important first step in elucidating gene function. Note that we could be surprised, however. Sometimes despite the knowledge we have accumulated about a gene, we find it works in an unexpected way – and produces a phenotype we would not have predicted. This is what is exciting about science- we don’t really know the answer until the experiments are done.
We will cover four means of eliminating or reducing gene expression here: insertional mutagenesis, homologous recombination, RNAi, and CRISPR-Cas9. There will be the strongest focus on the last two of these, both of which are borrowed from bacteria. We will consider how they work in nature, protecting cells from infection and then how they are used in analysis of gene function. The approaches used to make the constructs for these applications will also be described.
Contents
Learning Outcomes
Terminology before we begin
A. Insertional Inactivation
A-1. Ti Plasmids in Plants
A-2. P elements in fruit flies
C-1. A little history
C-2. How it works
C-3. Making constructs for RNAi
D-1. Preamble
D-2. How bacteria do it
D-3. How we do it: making CRISPR constructs
D3-i. Knock out – NHEJ repair
D3-ii. Directed modification – HDR
D3-iii. Designing oligonucleotides that will generate an overhang for cloning
Learning Outcomes
-
Describe the methods used to knock out or knock down gene function
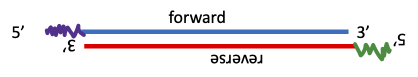
- Distinguish between forward and reverse genetic approaches
-
Distinguish between the techniques – used in different organisms, different approaches etc.
-
Describe how we make constructs for RNAi and how RNAi works
- Describe how to make a CRISPR construct through the method we will use in Bioinformatics Assignment#2
Terminology before we begin:
It is important to learn a bit of terminology before we begin. Forward genetics is a process by which we induce many mutations at random and then after we’ve done all the work of isolating the mutations, we have to still figure out which mutations we want to study, and then do a lot of analysis to find out which gene is mutated. Reverse genetics is an approach by which we identify what gene we want to study and target it specifically in some way. We then see what the effect is of altering the expression of the gene in order to determine what its function is. This has only been possible since the sequencing of the genomes of many model organisms. Both approaches have value but reverse genetics approaches are more targeted so we know exactly what changes we are making in the gene which helps us interpret the results. Forward genetics can produce mutations that you would never have designed on purpose and that produces an effect that is unexpected or unpredictable. But it is much more work than reverse genetics. We will be looking at one forward genetics approach and multiple reverse genetics approaches in this chapter.
Loss of function mutations are mutations that reduce or eliminate a gene’s function. In this case we may be eliminating the gene itself or preventing it from producing any of its protein product. These are examples of amorphic mutations; ones that are not producing any of the protein product they normally make. In other cases we might reduce the transcription of the gene but not completely eliminate it. Or a protein could be produced that is missing some amino acids or has some incorrect amino acids and though it is not very active, it is able to perform its function a little bit. These are examples of hypomorphic mutations, in these mutations the function of the gene is reduced but there is still some protein that somewhat does the job. To really understand the function of a gene we want to see what the phenotype of the amorphic mutation is but we like to use hypomorphic mutations for other purposes- both have their uses in genetics.
There are also gain of function mutations in which – for example – a gene is more active than usual either because it is being transcribed at a higher rate than usual, or because the protein can’t be degraded or down-regulated when it is supposed to stop performing its function. There may not be a phenotype associated with this type of hypermorphic mutation, but there could be a very strong and informative effect. It is very dependent on what the gene’s product does. Neomorphic mutations involve the mis-regulation of the gene or a change in the protein that causes it to do something different from the wild type situation. In these situations a gene might be expressed in a stage or type of tissue where it is not normally expressed. Or, the protein might be modified somehow so that it interacts with other proteins it would not normally interact with in a cell. Or it might localize to a place where it is not supposed to be. Sometimes these changes are very impactful. A signalling molecule that is active in the wrong tissue can promote cell division when it is not appropriate for the cells to be dividing- this could lead to tumour formation.
Click here for the powerpoint slides presented in the video below.
A. Insertional Mutagenesis:
One way to knock out a gene’s function is to make a mutation in it. There are various ways to cause changes in DNA sequence of genes. You can treat the organisms with chemicals or radiation and then look for the phenotypes that result. Then you must do a lot of work to figure out what gene has been mutated to cause the phenotype you see. This is an example of forward genetics. In this approach, mutations are induced (caused) at random and then you have to figure out what gene has been altered. A different way to make a mutation is to insert a segment of DNA into the gene. It is quite unlikely that adding a huge DNA sequence into a gene would leave the gene still functioning perfectly. This is because the gene will be frame-shifted. The gene can still be transcribed, but when the RNA is translated, at the point where the inserted DNA was transcribed, the amino acid sequence of the protein will be incorrect. And most likely there will be a stop codon in the sequence, leading to a truncated (shortened) protein that contains the wrong amino acids. This is called insertional inactivation and we have systems for using this approach in some of our commonly used model organisms. The insertion of the DNA into the genes to cause mutations is still random, but we can use our knowledge of the DNA we have introduced and the power of PCR to more easily determine which gene has been altered. We will talk about 2 types of insertional inactivation. The principles of these methods apply to other methods in other organisms you might learn about in other courses. (Note: For this semester, I am going to leave out the second part of this section, part A-2 about fruit flies)
A-1. Ti Plasmids in Plants
There is a type of soil dwelling bacterium that can infect plants and cause large “galls” which are a type of benign tumour. The tissues in these “galls” are reprogrammed by the bacterium to produce a protected place for the bacteria to live, and to also produce certain amino acids and other nutrients that the bacteria consume. The bacterium, Agrobacterium tumefaciens, contains a very large circular plasmid, 140 kb to 235 kb in size, called the Ti plasmid. The plasmid has features you would expect of any plasmid, such as an origin of replication. It also has a region called T-DNA which actually causes the formation of the tumour. During infection of the plant tissue, a copy of this T-DNA is inserted into the genome of the plant (the nuclear genome). In nature, this T-DNA has genes that encode enzymes that produce hormones in the host plant. It is this manipulation of the plant’s own hormones that leads to the formation of the tumour, called a crown gall.
There is also a region on the T-DNA responsible for synthesis of opines. These are unusual derivatives of sugars or amino acids and they provide nutrients to the bacteria living in the tumour tissue. The Ti plasmid also has genes on it that are needed for using the opines as a source of nutrition. These genes are not transferred to the plant. They are needed only by the bacteria.
The T-DNA has a short sequence (about 25 bp) to either side of it; these are called LB for left border and RB for right border. These sequences are necessary for the transfer of a copy of the T-DNA to the host plant’s genomic DNA. The Ti plasmid (but not the T-DNA) also has a virulence region; this is where the genes necessary to allow the bacterium to infect the host cell are found. There are some other regions on these huge plasmids that are not relevant for this topic. I am adding a simple diagram of the Ti plasmid from wikipedia. Pay attention to the LB and RB sequences, what is in the T-DNA part of the plasmid and what is not. The genes in the T-DNA segment are the ones that are copied and moved into the plant’s genome while the ones that are not in this region are functional in the bacterium only.

As with so many of our genetic engineering techniques, researchers have found something operating in nature and have figured out how to use it in research. The Ti plasmid has been modified for use in the genetic engineering of plants. We’ll talk later in the semester about some details of how the plasmid is used but for now the main thing to know is that one of the changes made is to remove the genes relating to plant hormone regulation and opine metabolism and to add a gene for antibiotic resistance. The plasmid can be introduced into plants using the antibiotic resistance as a selection mechanism. It is more complex than it sounds, and again, we will talk about the details of how the DNA gets into the plants and the process of selection etc. later in the semester. The result of transforming plant cells on a large scale is that many tens of thousands of lines of plants (perhaps by now, hundreds of thousands!) have been generated, each with a unique mutation in it, caused by an insertion of a large segment of T-DNA.
So far this is a lot like other forward genetics techniques but the work needed to try to figure out which gene has been altered by the mutation is much less than when genes are mutated by chemicals or radiation. This is because we know the exact DNA sequence of the T-DNA. And we can use this knowledge to design a primer to sequence the DNA of plants with a T-DNA insert into a gene. The primer recognizes the T-DNA sequence and is directed outward towards the gene sequence. This is a way of using the sequence we know – the T-DNA – to identify the sequence we don’t know – the plant gene.
Below is the recorded lecture for this topic (click here for the powerpoint slides):
A-2. P-elements in fruit flies
NOTE: for now, we will leave out this section. In many ways it is very similar to the plant example already explained. I’ve written it and it will stay in because it might be used in a later offering of the course. But you can ignore section A-2 this time.
In fruit flies, the transposable element called the P-element is used to create mutations and to make transgenic flies for other reasons. The P-element is remarkable: it is a gene which has the ability to move from place to place in the genome. The single gene on a P-element encodes transposase which is the name of the enzyme that cuts the DNA to either side of the element and inserts it into a new place in the genome.
P-elements were introduced into Drosophila melanogaster (the fruit fly most used in genetics research) probably no more than about 200 years ago. In the early days of fly research, flies isolated from the wild lacked P-elements and by the 1970 virtually every strain established from a wild population contained plenty of these elements. This is a tremendously rapid evolutionary change from the element being extremely rare in the 1920s to being practically ubiquitous by the 1970s.
It perhaps won’t surprise you to learn that P-elements have been modified from their wild form for use in fly research. For instance, the P-element has been transformed into a reporter construct, by putting a GFP gene on it, with the appropriate regulatory sequences and some of you who have taken BISC 302W already know quite a bit about all the interesting research being one with P-elements. Here we will just briefly outline their use in forward genetics approaches.
Like the Ti plasmid in plants, the P-element can be introduced into flies and allowed to insert into genes causing mutant phenotypes. In the fly community a concerted effort went into doing genetic screening on a large scale to try to produce a mutation in every single gene of the fly. Each mutation was of course kept as a separate line of flies that all have the same P-element induced mutation.
For genetic screening, the P-elements have had their transposase gene removed and replaced with the white+ gene. The + means that it is the wild type version of the gene. Fly genes are named backwards so the white+ gene is required to make the wild type red eye of the fly.
The P-element is introduced into flies by injecting a plasmid that contains the P-element (with its white+ reporter gene) into the posterior region of a very early embryo. At this stage the embryo is one big cell with many nuclei- the nuclei divide but the cell doesn’t. The posterior region is where we introduce the DNA because we are hoping it will be incorporated into a nucleus that goes on to form a germ cell. This will then give rise to gametes which may have the P-element somewhere in the genome. The injection is quite finicky- a very thin and very sharp needle is used to make the tiniest hole possible in the embryo. You don’t want to damage it. We inject the plasmid that contains the P-element and another plasmid that has the transposase gene on it. The transposase enzyme will cut the P-element out of the plasmid and insert it somewhere in the genome of some of the nuclei in the embryo. The flies we use for producing the embryos for injection are all mutant for the white gene. They have white eyes.
After the injection process we allow the embryos to recover and develop into adults. All the adults that develop have white eyes. That is because most won’t have a P-element in any of their cells. And those that do have the P-element incorporated into some of their cells won’t have it in the eyes, which develop from anterior cells in the embryo. The flies though are mated to either females or males that also have white eyes. Most of the progeny of these crosses have white eyes but some have red or orange or pinkish eyes. These flies developed from an gamete that had the P-element incorporated into the DNA of a germ cell. All of their cells now have the P-element in that position on that chromosome. And the white+ gene is being transcribed and translated from the P-element. If it is expressed a lot the eye is nearly wild type but if it is expressed just a little the eye might be just pinkish, very pale.
Once we have a P-element containing strain, we can do large scale genetic screens where we cause the element to mobilize randomly around the genome and we then collect a large number of individuals that have P element induced mutations. Each individual is used to make a strain of flies that all have the same mutation as the original fly. A consortium of researchers initiated a project to make a P-element mutation in every single gene of the fly as a resource for the community. There are thousands of strains available to researchers, each of which has a mutation in a single gene. And there are many P-elements, which have different components, for different purposes. For example, some have the lacZ gene as a reporter, while others have GFP; this is in addition to white+.
The value of having a P element induced mutation rather than chemical or radiation induced, is that it is fairly simple to figure out what gene the P-element is inserted into. There are two methods we might use, depending on which P-element is inserted into the gene.
The first method is called inverse PCR. It involves isolating the genomic DNA of individuals with the P-element mutation and cutting the DNA with an enzyme that cuts in a known location inside the P-element. The DNA is then ligated in a very large volume with a fairly small amount of DNA. We want the ligation mixture to be very dilute so that when the ligase glues two ends together they are most likely going to be the two ends of the same molecule. This generates a whole bunch of circles of DNA. A very few of these contain part of the P-element and a little bit of the sequence that was right beside the element. You set up a PCR reaction using a bit of the ligation mix and you use two primers that recognize the P-element and are directed outward toward the flanking DNA. A band is produced that can then be cloned into a plasmid, which is then used to transform cells. Colonies are grown overnight and plasmid minipreps are made by alkaline lysis. The DNA is then sent for sequencing. Even if you only have a small amount of DNA sequence it is usually enough to determine exactly where the element is located in the DNA of the fly.
Plasmid rescue is a quicker technique but is only possible if your mutation was caused by the right kind of P-element. Some have been designed that have a bacterial origin of replication and an ampicillin resistance gene on them. In this case you again isolate the genomic DNA of flies with the mutation, and you do a restriction digest of the DNA with a restriction enzyme that cuts in a particular location in the P-element. You also do the very dilute ligation. The usefulness of this type of P-element is that some of the circles that are made in this ligation will have some of the P-element DNA, some of the flanking fruit fly DNA, an amp resistance gene and an origin of replication. In essence you have made a little plasmid containing the DNA you want to sequence (the flanking DNA). So the PCR, cloning, etc. steps can be skipped. Instead you take your ligation mix and transform a small amount into some E. coli cells. You plate the transformed cells on amp plates and only those that contain the plasmid will survive. Then you can select colonies and grow some up and make plasmid preps from them, just as above.
B. Homologous Recombination:
In some organisms there is no convenient system for insertional mutagenesis. But we can use homologous recombination between the gene on the chromosome of a cell and an introduced construct to swap out the functional gene and replace it with a non-functional copy. In this case we know the exact sequence change that has been made to the gene and if we have removed the entire coding sequence we can be sure we have eliminated the gene function. The term recombination can refer both to crossing over and independent assortment. In this case we are referring specifically to crossover.
This requires a good understanding of the gene you are working with and knowledge of the sequence. The construct you design will contain segments of the DNA sequence to either side of the gene you want to knock out. But instead of the gene, you have an antibiotic resistance gene between the flanking sequences. Or in you might have a marker gene that produces a visible phenotype in individuals that have had their gene replaced by the marker. The image below will help explain the process. In this method we are taking advantage of the fact that crossing over can occur between two segments of DNA with homologous sequences. When two crossovers occur, the DNA between the two crossovers is “swapped”. In this way we can replace the actual gene we are studying, with the sequence we have placed on the construct. The vector, with the wild type copy of the gene “swapped onto” it, does not persist in the cells.
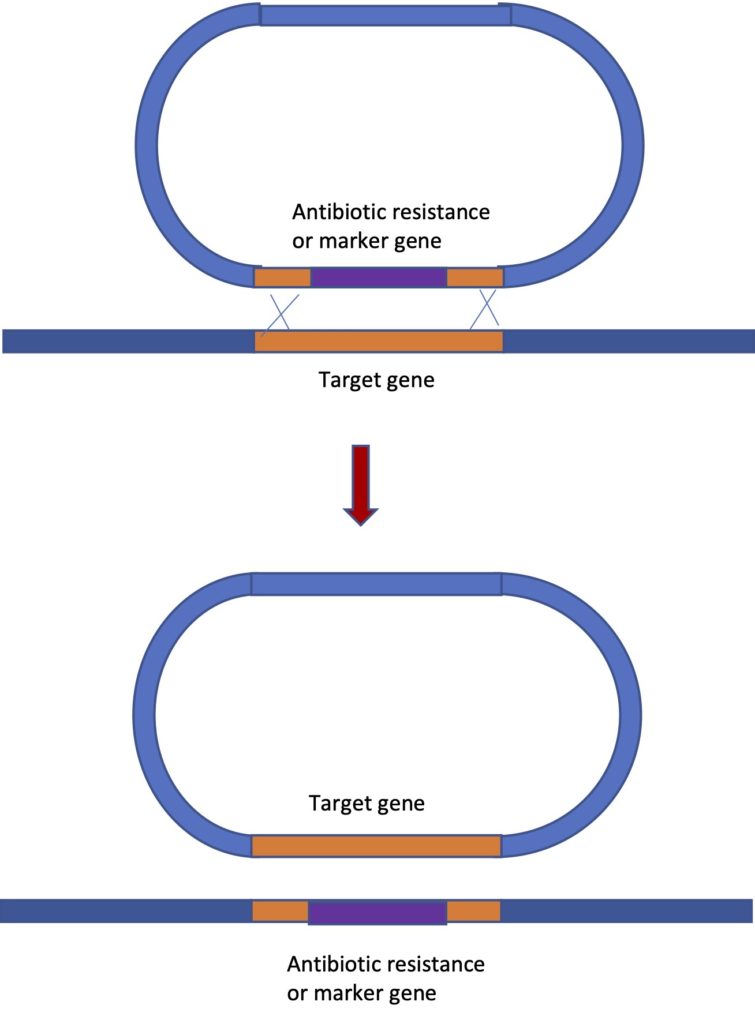
In the image, the vector is shown, with the antibiotic resistance gene (in purple) flanked by sequences to either side of the gene of interest. At least 2000 base pairs of flanking sequence is needed for this technique to work and many constructs that have been successfully used have quite a bit more- 6000 to 14000 nucleotides. Having a large amount of flanking sequence is needed to increase the chances of crossovers occurring. You need two crossovers to occur quite close to each other and this is a rare event. The process works well in many prokaryotes, in yeast, in mice and flies. But it does not work at all in plants or in human cells, which is unfortunate because if you think about it, this is a technique that could be used not only to replace a wild type (normal) copy of a gene with a different sequence in order to knock out the gene. It could do the reverse too – replace a mutated copy of a gene that is causing an inherited disease, with the wild type version – a form of gene therapy.
However, this is less disappointing than it used to be because we now have the CRISPR system of gene editing, which shows great potential for gene therapy and appears to work well in all organisms studied. The use of CRISPR for gene knockout will be described below and we may touch on it again towards the end of the semester when we talk about the use of genetic engineering in human medicine. The first self test question below is about this figure:

Click here for the powerpoint slides presented in the video below.
C. RNAi:
RNAi (RNA interference) is yet another process we use in genetic engineering that is borrowed from nature. It is a natural process among many eukaryotes. Once we understood how it works to reduce expression of a gene, we learned how to use it to perform targeted reduction of our genes of interest. And when I say “we” I specifically mean other people who figured this out. I am not one of those people.
C-1. A little history
I know this history very informally from the perspective of the fly research community. In the 1990s people were trying to knock out gene function by injecting anti-sense RNA into for example fly embryos, and then looking for phenotypes. The theory was that the antisense RNA would bind to the sense RNA in the cells and would prevent its translation. Thus we would get no functional protein and we could see the results of directly targeting a particular gene for inactivation. It was variably effective; sometimes it seemed to work well and sometimes less well. Then I was told at a meeting that someone figured out that the effect was stronger when both sense and antisense RNA was injected into the embryos. They had made a mistake by introducing both (by not linearizing the plasmid before transcribing the gene; see Chapter 7) and thought the experiment would not work at all. But it did and the effect was considerably stronger than just injecting the antisense RNA alone. This did not at first make much sense when we were talking about it then, but a few years later, the work of Fire and Mello, nematode researchers, showed that introducing double stranded RNA initiated a process that culminated in destruction of the sense RNA in a cell, via a process they called RNA interference. They published their work on this in 1998 and got the Nobel Prize for it in 2006. Of course, it turns out that plant researchers had also been circling around the same topic and had called it post-transcriptional gene silencing (PTGS). The first report of it in plants was published in 1990. So multiple research groups were figuring out the same process at around the same time.
The point about this method is that first off, somehow the introduction or production of double stranded RNA that corresponded to a particular gene led to the destruction of the normal mRNA from that gene and thus the gene’s function- the protein product – was eliminated or greatly reduced in amount. Second, the phenotype that results may tell you something about what the gene is needed for. This is an example of a loss of function mutation. Imagine there is a factory that builds wheelbarrows. There are 12 people that go into the factory every day and each does something to build the wheelbarrow. Everyone does their job. Nobody covers for anyone else. Now imagine that one of the people gets “inactivated” somehow- let’s imagine it is an impromptu vacation rather than something more sinister- and you are watching the wheelbarrows come out of the factory and they are missing the grips on the handles. What do you conclude? You can tell what the job of the absent worker was; their job was to install the part that is missing. This is very simplistic but gives an idea of how a loss of function mutation might suggest the function of a gene.
RNAi allows you to decide which gene to knock out, rather than trying to make random mutations and then find one in the gene you are studying. If you have done RNA in situ hybridization and/or made a reporter construct for your gene you will know where the gene is expressed and so you will know where to look for the phenotypes: the physical or physiological or health related etc. effects of reducing or eliminating the gene’s function.
C-2. How it works
RNAi is a process that works in most eukaryotes. It seems to be a defence mechanism against RNA viruses. Double stranded RNA is not normal in a cell so when it is detected, it is a sign that a retrovirus may have infected the cell. A protein called dicer, which acts as a homodimer (two identical subunits combine to perform the function) binds to double-stranded RNA in a non-sequence-specific way. That means it doesn’t recognize any particular sequence but just binds to any ds-RNA. Dicer is a type III RNA endonuclease. The size of the dimer dictates where the enzyme cuts the RNA. It cuts to either side of the complex leading to ~22 bp pieces of ds-RNA. The sizes vary slightly among organisms.
Then a complex called RISC (RNA Induced Silencing Complex) unwinds the ds-RNA pieces and hangs on to the antisense strand of the RNA. It uses this strand to target additional complementary RNA. When it finds a sequence that matches – the mRNA of the targeted gene- it cuts that RNA as well. The RNA endonuclease that does the cutting of the mRNA is called argonaut, but it is nicknamed “slicer”.
I am not clear on how the RISC complex hangs on to one strand of the RNA, the guide strand (which is the antisense strand) and lets go of the other strand, the passenger strand (the sense strand). But in any case, it does retain the antisense strand and this is how it is able to target the mRNA sequence of the gene.
There is a lot more to find out about the mechanism of RNAi if you are interested. It is also a mechanism that many organisms use to regulate their own genes. So some genes are transcribed but then at certain times in development, short anti-sense RNAs called microRNAs (miRNAs) bind to the mRNA and prevent its translation. They also target destruction of the message through dicer and the RISC complex because when they bind to the mRNA it creates ds-RNA, which triggers the RNAi process. It probably seems inefficient but it works. You will (or may have already) learn about it in Bisc 333.
There are human health connections as well. A form of macular degeneration is associated with an age-related down regulation of a form of dicer in the eye. There are sequences called Alu sequences in the human genome, and these are repetitive remnants of a retroviral infection in a human ancestor long ago. These are still expressed sometimes and are kept in check by miRNAs. When dicer is less expressed as the patient gets older, the Alu sequences begin to accumulate because there is a less efficient miRNA process to shut down their expression, and this accumulation leads to degeneration of the retina. I don’t know exactly how the accumulation of these sequences brings about the retinal degeneration, but it is interesting. Perhaps reintroducing active dicer through gene therapy could help treat this form of blindness.
C-3. Making constructs for RNAi
Constructs that will be used for RNAi don’t need to contain the entire sequence of the gene we are targeting, in fact it seems to work better if only part of the gene is used. A few hundred base pairs of sequence is usually enough. A standard length is 300 to 800 base pairs but for a gene I studied years ago, we used only about 80 base pairs because this was the largest section of the gene we could find that was unique, but I’ve since learned that many people have successfully used smaller sections of the gene they are targeting- even as small as 40 bp. It is probably somewhat related to the gene you are targeting. But whatever the size of your insert, the sequence you use must be unique to the gene you are studying. In the case of the gene I was interested in, there were two genes with very very similar sequence that performed slightly different functions in the flies during development. You might also be studying one member of a gene family and in this case many family members would have quite similar sequences. Sometimes the sequence chosen just has short stretches of sequence that are not unique to the gene we are targeting. You only need 22 nucleotides of such sequence to generate “off-target” effects. This means that the RNAi process will not only inactivate mRNA from the intended gene but also for another gene or genes which are not the intended target. You don’t want the phenotype you see to result from knocking out multiple genes because you will not know this and will assume that the cool phenotype you observe is the result of knocking down or knocking out your intended gene. You can search the sequence you plan to use in your construct for short regions that match different genes than the intended one. If you find such matches, a different section of the gene must be used.
The construct you make must produce double stranded RNA. So the construct needs a promoter to transcribe the DNA that you insert into the vector. You can insert the same DNA sequence twice into the same vector, but in opposite orientation. The RNA polymerase will transcribe the first copy and will continue through the second copy- because this copy is in the opposite orientation, the RNA of the second half of the transcript will be the reverse complement of the first half. Therefore it can fold back on itself to product ds-RNA. We often include a short spacer between the two inserts to make the folding of the RNA a bit easier. Vectors have been designed with two cloning sites that can allow you to automatically put your inserts into the vector in the correct orientation. We will talk about this “recombinase” cloning method later in in the semester. We can use restriction enzyme cloning too, but it is a lot of work- we have to clone one insert into the vector at a time and we have to use different restriction enzymes for the two inserts so that we are only affecting one insert at a time. And! We always have to be really careful in planning our cloning procedure to ensure we have the inserts in opposite orientation. Below is an image of just part of such a construct, showing the two inserts which are the exact same sequence but in opposite orientation, and the promoter to one side and terminator to the other. The spacer between is to help the formation of a hairpin- double stranded RNA with a loop at one end. The loop may be degraded after the RNA folds into the hairpin structure.
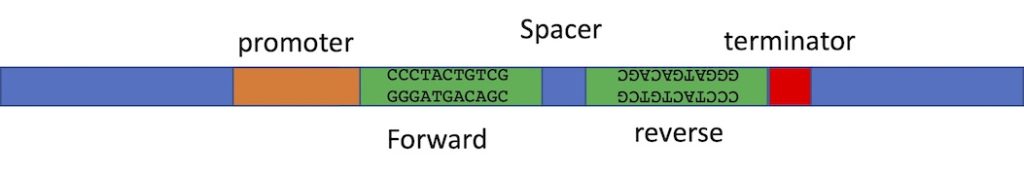
The second approach is much easier. In this case your insert is cloned into the polylinker and there are two copies of the same promoter- one to either side of your insert. Once you’ve made the transgenic organism, the insert can be transcribed in both directions at the same time, making two complementary RNAs that will bind each other to become ds-RNA. The image below shows the two promoters to either side of the insert which can be in any orientation.

RNAi can be done in a transient way as is often done using cultured cells. The double stranded RNA is made in vitro and then isolated and injected into the cells or into embryos (this is done in fly embryos sometimes). Or a plasmid is introduced that will transcribe the ds-RNA but the plasmid is not stable in the cells, and so will be degraded in a relatively short time. These approaches lead to only a transient assay because you introduce the RNA, and you then have a few hours to see the effect. It could be a change in cell behaviour or in gene expression or some other immediate result of the RNAi. After a few hours the effect diminishes as all the ds-RNA has been cut up and used in the response and/or the plasmid has been degraded by the cell.
To do a longer term experiment the construct you’ve made to produce the ds-RNA is introduced into an organism and part of the plasmid is integrated into the genome of the organism. Then a strain or line of that organism that contains the construct is kept. We generally like to use inducible promoters in that case, so that we can turn on the production of ds-RNA at a defined time or place and observe the effects. In some organisms, we can use a promoter called the heat-shock promoter. Heat shock proteins are chaperones that help keep proteins from unfolding during high temperature or other types of stress in a cell. The promoters of these proteins are very strong and they induce a large amount of expression. The RNA polymerases and some other regulatory proteins are sitting on these promoters all the time, ready to start transcription the moment these proteins are needed- which by definition is going to be some type of emergency situation for the cell. So when there is a sudden temperature increase the heat shock genes are transcribed vigorously to produce lots of chaperone proteins and protect the cell from the damaging effects of the heat treatment. When we attach these promoters to other genes they will be highly transcribed under our control- when we heat shock the cells or organisms that carry the construct. In different systems, different inducible promoters may be used, but the point is the same- we can turn on the RNAi at specific times to see the effect of knocking out a gene at a particular time.
Sometimes the RNAi effect is not very strong. In that case you can sometimes introduce two different RNAi constructs into the organism, each targeting a different part of the gene. That can increase the effect. Also, you can introduce a construct that has extra copies of the dicer gene on it. Extra copies of the dicer gene mean more of the dicer protein and the construct should have dicer under the control of the same inducible promoter as your RNAi construct. So, when you induce the ds-RNAi for your gene of interest, at the same time you make a lot of extra dicer protein and this greatly enhances the RNAi response.
Click here for the powerpoint slides presented in the video below.
D. CRISPR:
D-1. Preamble
A few days ago the 2020 Nobel Prize for Chemistry was awarded to Jennifer Doudna and Emmanuelle Charpentier for their work on the CRISPR-Cas 9 system. They were studying how bacteria can protect themselves from a second viral infection after surviving the first infection and this curiosity-driven research led to the elucidation of a mechanism by which we could edit genes using the same system. Please read the following short article that describes a bit about the research and the researchers. It also contains a short scrollable description of how the whole process works that is easy to understand and nicely illustrated. Your next bioinformatics project is the production of oligonucleotides to make a CRISPR construct for a gene in the foxtail millet, Setaria viridis. This will contribute toward a global research project that I will tell you about next week.
The Conversation: What is CRISPR, the gene editing technology that won the Chemistry Nobel prize?
D-2. How bacteria do it
Bacteria that survive an infection by a bacteriophage (virus that infects bacteria) cut out and keep a small segment of the phage DNA and store it in a region of the bacterial chromosome that contains a set of short repeats. Between the repeats are the DNA records of previous viral infections- segments of viral DNA. The repeats are called Clustered Regularly Interspaced Short Palindromic Repeats. This is where the term CRISPR comes from. The DNA from the phage that is stored in the CRISPR sites is not just random sequence; it is very precise: it is 20 nucleotides of sequence that are found immediately upstream of a sequence: NGG. This is called a PAM sequence, which means protospacer adjacent motif. The NGG sequence is not inserted in the CRISPR loci but it is in the corresponding phage DNA and is important for the bacterial response to infection later.
When bacteria are infected again by the same type of phage – not the same bacterium but the descendants of the cell that survived the infection – the CRISPR loci are transcribed to make RNA. The RNA that comes from the previous phage DNA is the crRNA. Another RNA is also transcribed, which is called the tracrRNA. These are functional RNAs; they are not translated into protein but perform their functions as RNAs and they have regions where the two RNAs are complementary to each other and so can base pair with each other. A third gene is also transcribed and translated to produce the Cas9 protein. Cas means CRISPR Associated protein 9. There are many of these in different systems but we’ll stick to Cas9 for BISC 357. (It has had other names in the past: Cas5, Csn1, or Csx12 so if you come across these in your studies or in other courses, you will know that these also refer to the Cas9 protein)
So the bacteria are undergoing an infection and there are two RNAs and one protein produced in response. The tracrRNA has a region that binds to the Cas9 protein and the region of complementarity to the crRNA. It acts as a linker between the two. The crRNA is complementary to one strand of the phage DNA. So it binds to the phage DNA and the tracrRNA forms the link connecting the Cas9 protein to the crRNA and when Cas9 is in position on the phage DNA it cuts the phage DNA in a very precise location- 3 nucleotides upstream of the PAM sequence. It cuts both strands of the DNA to make a “break”. The phage gene cannot be expressed and furthermore, the DNA is degraded by the bacterial exonucleases. This is where the PAM sequence is essential. The PAM sequence is unique to each type of Cas protein and is absolutely required for the protein to be able to cut the DNA. So even if the crRNA recognizes the 20 bases of target sequence in the phage genome and the tracrRNA connects the Cas9 protein to the crRNA, Cas9 must bind to the PAM sequence or it will be unable to undergo the conformational shift that activates the endonuclease function.
Bacteria are really quite remarkable.
Click here for the powerpoint slides presented in the video below.
D-3. How we use it: the principle of gene editing
When we make constructs for CRISPR we use vectors that have everything we need except for the unique 20 bases of sequence that will target the gene we want to knock out. The gene for Cas9 is on the vector because the organisms we are studying don’t have this protein. The gene for Cas9 has been modified to contain one or more nuclear localization signals (NLSs). The protein must enter the nucleus to cut the target DNA so the NLS is required to direct the protein to the correct location to do its job. Bacteria have no nuclear envelope and so do not require NLSs on their Cas9 protein. The two genes for tracr RNA and crRNA have been fused. The “overlap” region is removed and the two parts of the RNA that are needed to direct Cas9 to the target DNA are transcribed as a single guide RNA (sgRNA). At one end is the 20 nucleotide sequence that is designed by the researcher and is specific for the gene of interest. The part that folds up and binds with Cas9 is the same for every gene, so this is an unvarying part of the construct. Both the Cas9 gene and the sgRNA gene are under the control of strong promoters that work in eukaryotes.
The constructs are introduced into organisms in a variety of ways, depending on the organism. Once inside the single guide RNA is expressed as is the Cas9 protein. They enter the nucleus and the sgRNA binds the target sequence on the gene of interest. The Cas9 protein interacts with the PAM sequence and changes conformation in order to cut the DNA 3 bases upstream of the NGG sequence.
Once a cut is made in the DNA sequence, several things can happen. An accurate repair system might repair the DNA. If this happens it might be cut again. This may happen repeatedly. But there are repair systems that are less accurate that can “repair” the DNA but that will introduce mistakes in the sequence in the process. These are the ones that will inactivate the gene.
Click here for the powerpoint slides presented in the video below.
D3-i. Knock out – NHEJ repair
When the non-homologous end joining (NHEJ) repair system acts, it reconnects broken DNA but not in an “accurate” way. This type of repair can cause insertion or deletions into the DNA at the site of the break and if multiple breaks occur along a chromosome, the “wrong” ends might be connected, to generate inversions or translocations. I always think of this repair system as the “Emergency! Just glue the pieces back together, don’t worry about getting it perfect!” type of system. So in the case that we introduce our construct that targets our gene, in many cells we expect that the NHEJ repair system may repair the breaks that are induced in the gene. Consider that if even one nucleotide is added at the break, the entire gene is frame-shifted. Thus the correct protein won’t be produced. Or if nucleotides are deleted and they are not in multiples of three we will also get a frameshift mutation. The mutations are called “indel” because either insertion or deletion of one or two nucleotides may occur and in both situations, a frameshift results. Frameshifts usually result in truncated proteins- you may recall from your gene annotation assignments, that all reading frames except the correct one contain lots of stop codons.
Then we can look at the phenotype of the gene knockout to see the consequences of inactivating the gene. We will probably also collect DNA from the organism and sequence the gene to determine what the exact change in the sequence was. We may obtain several individuals with mutant phenotypes; they may not have exactly the same genetic change.
D3-ii. Directed modification – HDR
Homology dependent repair is accurate and sequence-based. If a break occurs in the DNA, proteins bind it, and generate single stranded sections on the ends of the break that then do a “homology search”. The strands generally find the homologue (we are talking about diploid organisms here) and use that homologue to copy the correct sequence into the broken region.
In general this repair system would not result in a mutation of the gene we are targeting. However, if we provide some “donor” DNA along with the construct that we introduce into the research organism, we are essentially providing the homology dependent repair systems a piece of DNA to copy into the broken region. In this case we could introduce a reporter gene, or a piece of DNA with a particular type of mutation that we want to study. Suppose we thought a certain set of a few amino acids were the critical ones for a protein’s function. We could provide a donor DNA sequence that lacks the codons for those amino acids, and the repair system could incorporate that into the gene. We could then check to see whether we were correct about the protein’s function.
Click here for the powerpoint slides presented in the video below.
There is a former student from 357 who also TAd the course a couple of years ago who is currently using this very approach to study heart disease in a zebra fish model for his PhD research.
Perhaps you’ve already thought of the most interesting application of this approach: the possibility of cutting a target DNA sequence that is actually causing a disease or condition in a person, and replacing that mutated sequence with the wild type form, in order to treat the condition. This has already been tried with sickle cell anemia, and within the last few years, there were reports of a person who had been treated for sickle cell anemia using CRISPR. In sickle cell anemia the hemoglobin gene has a mutation in it that causes the hemoglobin molecules to bind incorrectly to each other. They don’t carry oxygen as effectively as regular hemoglobin and they cause a deformation of the red bloods cells that causes the cells to get stuck in small capillaries sometimes. This can cause severe pain and because the oxygen carrying capacity is low, people with this condition have low energy. To treat this condition, stem cells were collected from the woman with the condition. The CRISPR treatment was performed on the cells to inactivate a gene called BCL11A. This gene shuts off production of fetal hemoglobin a few months after a baby is born. Inactivating this gene in the adult woman’s stem cells allowed the fetal hemoglobin gene to turn back on in those cells. The woman underwent a round of chemotherapy, presumably to kill the stem cells in her bone marrow and then the CRISPR-treated stem cells were reintroduced into her body. Producing large amounts of the fetal hemoglobin molecule (which is wild type) prevents the sickling of the blood cells. As a bonus, the fetal form of hemoglobin has higher oxygen carrying capacity than the adult form. The person who was treated seems to be in good health, a couple of years after the treatment. There is a short article about this, from July of 2022 linked below:
healthline: First Person Treated for Sickle Cell Disease with CRISPR Is Doing Well
D3-iii. Designing oligonucleotides that will generate an overhang for cloning
This is not specific to CRISPR, but is an approach we are going to use in our CRISPR bioinformatics exercise, so it is a good place to introduce it. We design oligonucleotides that are complementary to each other, EXCEPT for four nucleotides at the 5′ ends of the oligos. We are going to clone into a vector cut with a special enzyme which cuts OUTSIDE of the recognition sequence so that the overhang generated is different at each site, depending on what sequence is near the recognition site. Our vector has two restriction sites (BsaI) and the overhangs generated are not complementary to each other. Thus the vector cannot re-circularize during ligation.
To make the insert, we take equimolar amounts (the same # of molecules) of each oligo and combine them in a PCR tube. We heat to 96C for about 5 minutes and then allow the temperature to decrease very gradually. This allows the complementary parts of the forward and reverse oligos to find each other and to bind. The 5′ ends stick out (see below) and the sequences that are sticking out are complementary to the overhangs in the vector. There is more information in the lecture on CRISPR, the bioinformatics assignment in which you design the oligos for this project, the lecture about the project we’re working on, as well as your lab handout, later in the semester. In the image below, the squiggly lines at the 5′ ends of the annealed oligos are the 4-bp sequences that match the insertion site in the cut vector.